SEA-AD Multiregion taxonomy and snRNASeq analysis: clustering and annotations#
Alzheimer’s disease (AD) is characterized by the progressive accumulation of amyloid-beta (Aβ) plaques and hyperphosphorylated tau (pTau) tangles across brain regions. While these regions differ in architecture, function, and susceptibility to pathology, many share common cellular populations. The SEA-AD Multiregion Taxonomy was developed to provide a unified cellular framework for studying cellular vulnerability and molecular change across the full arc of canonical AD progression.
This taxonomy is derived from a multimodal dataset generated from 84 deeply characterized donors spanning the full spectrum of AD pathology. The dataset presented here contains approximately 6 million high-quality nuclei from single-nucleus RNA-seq and Multiome profiling, along with an additional 2.6 million nuclei that did not pass quality-control criteria. These data were generated alongside approximately 1 million single-nucleus ATAC-seq profiles, which informed multimodal analyses but are not included in the resources described here. Single-nucleus profiling was performed across ten brain regions: medial entorhinal cortex (MEC), lateral entorhinal cortex (LEC), hippocampus (HIP), inferior temporal gyrus (ITG), middle temporal gyrus (MTG), superior temporal gyrus (STG), dorsolateral prefrontal cortex (BA9), frontal insula (FI), angular gyrus (AnG), and primary visual cortex (V1C). Nuclei were mapped to an expanded reference taxonomy comprising 207 cell types, organized into 28 subclasses and 3 major cellular classes, including both broadly shared and regionally specialized populations. Cell type names are harmonized between studies; for example, Sst 25 from Gabitto, Travaglini et al. (2024) and Sst 25 in this taxonomy refer to the same cell population.
To generate this taxonomy, nuclei were subjected to standardized quality control, integrated across regions and modalities, and hierarchically mapped using deep-learning approaches based on scVI and scANVI. The resulting taxonomy provides consistent cell-type nomenclature across SEA-AD resources while preserving region-specific cellular specializations found in allocortical and neocortical regions. Spatial transcriptomic datasets were additionally used to anatomically localize newly defined excitatory populations within the hippocampus and entorhinal cortex.
The notebook presented here demonstrates how to access and visualize taxonomy annotations, cell type metadata, donor information, brain region annotations, and AD-associated cellular changes from the SEA-AD multiregional dataset. These examples focus on precomputed metadata accessible through the Allen Brain Cell Atlas Access package. Additional data, including links to raw sequencing, spatial transcriptomic, and neuropathology data, are available at SEA-AD.org.
%matplotlib inline
import pandas as pd
from pathlib import Path
import numpy as np
import matplotlib.pyplot as plt
from typing import Tuple, Optional
from abc_atlas_access.abc_atlas_cache.abc_project_cache import AbcProjectCache
We will interact with the data using the AbcProjectCache. This cache object downloads data requested by the user, tracks which files have already been downloaded to your local system, and serves the path to the requested data on disk. For metadata, the cache can also directly serve up a Pandas DataFrame. See the getting_started notebook for more details on using the cache including installing it if it has not already been.
Change the download_base variable to where you would like to download the data in your system or a location where a cache is already available.
download_base = Path('../../data/allen-brain-cell-atlas-staging/')
abc_cache = AbcProjectCache.from_cache_dir(
download_base
)
abc_cache.current_manifest
'releases/20260711/manifest.json'
Data overview#
Below we list the metadata and gene expression files for each of the directories that make up this dataset:
SEA-AD-Multiregion-10X
SEA-AD-Multiregion-taxonomy
print("SEA-AD-10X: gene expression data (h5ad)\n\t", abc_cache.list_expression_matrix_files(directory='SEA-AD-Multiregion-10X'))
print("SEA-AD-Multiregion-10X: metadata (csv)\n\t", abc_cache.list_metadata_files(directory='SEA-AD-Multiregion-10X'))
SEA-AD-10X: gene expression data (h5ad)
['AnG-10X/log2', 'AnG-10X/raw', 'DFC-10X/log2', 'DFC-10X/raw', 'FI-10X/log2', 'FI-10X/raw', 'HIP-10X/log2', 'HIP-10X/raw', 'ITG-10X/log2', 'ITG-10X/raw', 'LEC-10X/log2', 'LEC-10X/raw', 'MEC-10X/log2', 'MEC-10X/raw', 'MTG-10X/log2', 'MTG-10X/raw', 'STG-10X/log2', 'STG-10X/raw', 'V1C-10X/log2', 'V1C-10X/raw']
SEA-AD-Multiregion-10X: metadata (csv)
['cell_metadata', 'disease', 'donor', 'example_gene_expression', 'gene', 'library', 'value_sets']
We will also use metadata from the SEA-AD-Multiregion-taxonomy directory. Below is the list of available files:
print("SEA-AD-Multiregion-taxonomy: metadata (csv)\n\t", abc_cache.list_metadata_files(directory='SEA-AD-Multiregion-taxonomy'))
SEA-AD-Multiregion-taxonomy: metadata (csv)
['cell_2d_embedding_coordinates', 'cell_to_cluster_membership', 'cluster', 'cluster_annotation_term', 'cluster_annotation_term_set', 'cluster_to_cluster_annotation_membership']
Cell metadata#
Essential cell metadata is stored as a CSV file that we load as a Pandas DataFrame. Each row represents one cell indexed by a cell label. The cell label is the concatenation of barcode and name of the sample. In this context, the sample is the barcoded cell sample that represents a single load into one port of the 10x Chromium. Note that cell barcodes are only unique within a single barcoded cell sample and that the same barcode can be reused exp_component_name is an alternative cell identifier that indexes some of the data hosted at SEA-AD.org.
Each cell is associated with a library label, donor label, alignment_job_id, feature_matrix_label and dataset_label identifying which data package this cell is part of. This metadata file will be combined with other metadata files that come with this package to add information associated with the donor, UMAP coordinates, taxonomy assignments, and more.
Below, we load the first of the metadata used in this tutorial. This represents the cell metadata for the aligned dataset.
The command we use below both downloads the data if it is not already available in the local cache and loads the data as a Pandas DataFrame. This pattern of loading metadata is repeated throughout the tutorials.
cell = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-10X',
file_name='cell_metadata'
).set_index('cell_label')
print("Number of cells = ", len(cell))
cell.head()
cell_metadata.csv: 100%|██████████| 1.64G/1.64G [02:44<00:00, 9.98MMB/s]
/Users/chris.morrison/src/abc_atlas_access/src/abc_atlas_access/abc_atlas_cache/abc_project_cache.py:643: DtypeWarning: Columns (4) have mixed types. Specify dtype option on import or set low_memory=False.
return pd.read_csv(path, **kwargs)
Number of cells = 8641342
| cell_barcode | barcoded_cell_sample_label | library_label | alignment_job_id | doublet_score | umi_count | donor_label | exp_component_name | feature_matrix_label | dataset_label | |
|---|---|---|---|---|---|---|---|---|---|---|
| cell_label | ||||||||||
| AAACAGCCACTGGCTG-2001_A08 | AAACAGCCACTGGCTG | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.184615 | 54020.0 | H21.33.001 | AAACAGCCACTGGCTG-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X |
| AAACAGCCAGGTTATT-2001_A08 | AAACAGCCAGGTTATT | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.060606 | 4438.0 | H21.33.001 | AAACAGCCAGGTTATT-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X |
| AAACAGCCATTCCTCG-2001_A08 | AAACAGCCATTCCTCG | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.276923 | 66285.0 | H21.33.001 | AAACAGCCATTCCTCG-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X |
| AAACATGCAATGCCTA-2001_A08 | AAACATGCAATGCCTA | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.060606 | 6019.0 | H21.33.001 | AAACATGCAATGCCTA-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X |
| AAACATGCACCTCACC-2001_A08 | AAACATGCACCTCACC | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.181818 | 48653.0 | H21.33.001 | AAACATGCACCTCACC-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X |
We can use pandas groupby function to see how many unique items are associated for each field and list them out if the number of unique items is small.
def print_column_info(df):
for c in df.columns:
grouped = df[[c]].groupby(c).count()
members = ''
if len(grouped) < 30:
members = str(list(grouped.index))
print("Number of unique %s = %d %s" % (c, len(grouped), members))
print_column_info(cell)
Number of unique cell_barcode = 3767416
Number of unique barcoded_cell_sample_label = 908
Number of unique library_label = 908
Number of unique alignment_job_id = 913
Number of unique doublet_score = 2792
Number of unique umi_count = 177044
Number of unique donor_label = 84
Number of unique exp_component_name = 8641342
Number of unique feature_matrix_label = 10 ['AnG-10X', 'DFC-10X', 'FI-10X', 'HIP-10X', 'ITG-10X', 'LEC-10X', 'MEC-10X', 'MTG-10X', 'STG-10X', 'V1C-10X']
Number of unique dataset_label = 1 ['SEA-AD-Multiregion-10X']
Donor, Library, and Disease metadata#
The first two associated metadata we load are the donor, library, and disease tables. The donor table includes donor demographics such as species, age, sex, race, and education, along with brain tissue metrics such as pH, postmortem interval (PMI), and fresh brain weight. This table also indicates which study a donor was enrolled in (ACT or ADRC). The library table contains information on 10X methods and brain region of interest the tissue was extracted from. The disease table contains disease progression metrics and other pathology data. Definitions of many of these columns can be found here.
donor = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-10X',
file_name='donor',
).set_index('donor_label')
donor
donor.csv: 100%|██████████| 17.1k/17.1k [00:00<00:00, 235kMB/s]
| donor_species | species_scientific_name | species_genus | donor_sex | donor_gender | donor_age | donor_age_value | donor_age_unit | Race (choice=White) | Race (choice=Black/ African American) | ... | Race (choice=Other) | Hispanic/Latino | specify other race | Highest level of education | Years of education | PMI | Fresh Brain Weight | Brain pH | Primary Study Name | donor_race | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| donor_label | |||||||||||||||||||||
| H19.33.004 | NCBITaxon:9606 | Homo sapiens | Human | Female | Female | 80 yrs | 80 | years | Checked | Unchecked | ... | Unchecked | No | NaN | Bachelors | 17 | 8.133333 | 1035.0 | 7.0 | ACT | White |
| H20.33.001 | NCBITaxon:9606 | Homo sapiens | Human | Male | Male | 82 yrs | 82 | years | Checked | Unchecked | ... | Unchecked | No | NaN | Bachelors | 16 | 7.700000 | 1338.0 | 6.8 | ACT | White |
| H20.33.002 | NCBITaxon:9606 | Homo sapiens | Human | Female | Female | 97 yrs | 97 | years | Checked | Unchecked | ... | Unchecked | No | NaN | High School | 12 | 4.333333 | 1078.0 | 7.3 | ACT | White |
| H20.33.004 | NCBITaxon:9606 | Homo sapiens | Human | Male | Male | 86 yrs | 86 | years | Checked | Unchecked | ... | Unchecked | No | NaN | Trade School/ Tech School | 15 | 8.833333 | 1261.0 | 6.7 | ACT | White |
| H20.33.005 | NCBITaxon:9606 | Homo sapiens | Human | Female | Female | 99 yrs | 99 | years | Checked | Unchecked | ... | Unchecked | No | NaN | High School | 12 | 7.600000 | 1003.0 | 6.8 | ACT | White |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| H21.33.043 | NCBITaxon:9606 | Homo sapiens | Human | Female | Female | 95 yrs | 95 | years | Checked | Unchecked | ... | Unchecked | No | NaN | Bachelors | 16 | 4.400000 | 1082.0 | 6.6 | ACT | White |
| H21.33.044 | NCBITaxon:9606 | Homo sapiens | Human | Female | Female | 88 yrs | 88 | years | Checked | Unchecked | ... | Unchecked | No | NaN | Trade School/ Tech School | 15 | 7.000000 | 1168.0 | 6.6 | ACT | White |
| H21.33.045 | NCBITaxon:9606 | Homo sapiens | Human | Female | Female | 94 yrs | 94 | years | Unchecked | Unchecked | ... | Unchecked | No | NaN | High School | 12 | 4.000000 | 925.0 | 7.2 | ADRC Clinical Core | Asian |
| H21.33.046 | NCBITaxon:9606 | Homo sapiens | Human | Male | Male | 97 yrs | 97 | years | Checked | Unchecked | ... | Unchecked | No | NaN | Professional | 17 | 7.000000 | 1159.0 | 6.4 | ACT | White |
| H21.33.047 | NCBITaxon:9606 | Homo sapiens | Human | Female | Male | 90 yrs | 90 | years | Checked | Unchecked | ... | Unchecked | No | NaN | Professional | 21 | 4.400000 | 1168.0 | 7.2 | ACT | White |
84 rows × 24 columns
Next we load the library metadata. The information we will primarily use from this table are the region of interest that each library is associated with
library = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-10X',
file_name='library'
).set_index('library_label')
library
library.csv: 100%|██████████| 117k/117k [00:00<00:00, 564kMB/s]
| library_method | barcoded_cell_sample_label | enrichment_population | cell_specimen_type | region_of_interest_label | region_of_interest_name | parcellation_term_identifier | Brain Region | donor_label | |
|---|---|---|---|---|---|---|---|---|---|
| library_label | |||||||||
| L8XR_231221_02_D02 | 10xMultiome;GEX | 2001_A08 | 70% NeuN+, 30% NeuN- | Nuclei | AnG | angular gyrus | DHBA:12136 | AnG | H21.33.001 |
| L8XR_231130_21_B02 | 10xMultiome;GEX | 1963_A06 | 70% NeuN+, 30% NeuN- | Nuclei | AnG | angular gyrus | DHBA:12136 | AnG | H21.33.002 |
| L8XR_231116_02_B08 | 10xMultiome;GEX | 1957_A08 | 70% NeuN+, 30% NeuN- | Nuclei | AnG | angular gyrus | DHBA:12136 | AnG | H20.33.035 |
| L8XR_231130_21_E02 | 10xMultiome;GEX | 1967_B09 | 70% NeuN+, 30% NeuN- | Nuclei | AnG | angular gyrus | DHBA:12136 | AnG | H21.33.028 |
| L8XR_231221_02_G02 | 10xMultiome;GEX | 2001_C08 | 70% NeuN+, 30% NeuN- | Nuclei | AnG | angular gyrus | DHBA:12136 | AnG | H21.33.004 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| L8XR_240530_01_G08 | 10xMultiome;GEX | 2252_A07 | 70% NeuN+, 30% NeuN- | Nuclei | FI | frontal agranular insular cortex (area FI) | DHBA:10329 | FI | H20.33.018 |
| L8XR_240607_02_A04 | 10xMultiome;GEX | 2252_B07 | 70% NeuN+, 30% NeuN- | Nuclei | FI | frontal agranular insular cortex (area FI) | DHBA:10329 | FI | H20.33.028 |
| L8XR_240607_02_F04 | 10xMultiome;GEX | 2252_C07 | 70% NeuN+, 30% NeuN- | Nuclei | FI | frontal agranular insular cortex (area FI) | DHBA:10329 | FI | H20.33.033 |
| L8XR_240620_01_F09 | 10xMultiome;GEX | 2284_C09 | 70% NeuN+, 30% NeuN- | Nuclei | FI | frontal agranular insular cortex (area FI) | DHBA:10329 | FI | H20.33.037 |
| L8XR_240801_02_F09 | 10xMultiome;GEX | 2351_B09 | 70% NeuN+, 30% NeuN- | Nuclei | FI | frontal agranular insular cortex (area FI) | DHBA:10329 | FI | H21.33.026 |
908 rows × 9 columns
Finally we load the disease data containing disease progression information.
disease = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-10X',
file_name='disease'
).set_index('donor_label')
disease
disease.csv: 100%|██████████| 14.4k/14.4k [00:00<00:00, 94.3kMB/s]
| Overall AD neuropathological Change | Thal | Braak | CERAD score | Overall CAA Score | Highest Lewy Body Disease | Total Microinfarcts (not observed grossly) | Total microinfarcts in screening sections | Atherosclerosis | Arteriolosclerosis | ... | Cognitive Status | Last CASI Score | Interval from last CASI in months | Last MMSE Score | Interval from last MMSE in months | Last MOCA Score | Interval from last MOCA in months | APOE Genotype | Severely Affected Donor | CPS_Global | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| donor_label | |||||||||||||||||||||
| H19.33.004 | Not AD | Thal 0 | Braak IV | Absent | Not identified | Not Identified (olfactory bulb not assessed) | 1 | 1 | Mild | Moderate | ... | No dementia | 85.0 | 3.5 | 25.0 | 3.5 | NaN | NaN | 3/3 | N | 0.102055 |
| H20.33.001 | Low | Thal 2 | Braak IV | Sparse | Not identified | Not Identified (olfactory bulb not assessed) | 0 | 0 | Mild | Mild | ... | No dementia | 97.0 | 18.2 | 28.0 | 18.2 | NaN | NaN | 3/3 | N | 0.390107 |
| H20.33.002 | Not AD | Thal 0 | Braak IV | Absent | Not identified | Limbic (Transitional) | 0 | 0 | Moderate | Moderate | ... | No dementia | 93.0 | 46.1 | 33.0 | 22.6 | NaN | NaN | 2/3 | N | 0.081825 |
| H20.33.004 | High | Thal 5 | Braak V | Frequent | Moderate | Neocortical (Diffuse) | 0 | 0 | Mild | Severe | ... | Dementia | 80.0 | 55.7 | 25.0 | 55.7 | NaN | NaN | 3/4 | N | 0.754141 |
| H20.33.005 | Intermediate | Thal 3 | Braak IV | Moderate | Moderate | Not Identified (olfactory bulb not assessed) | 2 | 2 | Mild | Moderate | ... | No dementia | 94.0 | 24.2 | 29.0 | 24.2 | NaN | NaN | 2/3 | N | 0.276006 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| H21.33.043 | Low | Thal 4 | Braak II | Sparse | Not identified | Not Identified (olfactory bulb assessed) | 1 | 0 | Moderate | Moderate | ... | Dementia | 97.0 | 35.1 | 29.0 | 35.1 | NaN | NaN | 3/3 | N | 0.580067 |
| H21.33.044 | Intermediate | Thal 3 | Braak VI | Frequent | Moderate | Not Identified (olfactory bulb not assessed) | 9 | 9 | Mild | Severe | ... | Dementia | 81.0 | 8.9 | 21.0 | 8.9 | NaN | NaN | 3/3 | N | 0.731159 |
| H21.33.045 | High | Thal 4 | Braak VI | Frequent | Moderate | Limbic (Transitional) | 0 | 0 | Moderate | Moderate | ... | Dementia | NaN | NaN | 17.0 | 65.3 | NaN | NaN | 3/4 | Y | 0.962116 |
| H21.33.046 | High | Thal 4 | Braak V | Moderate | Moderate | Neocortical (Diffuse) | 0 | 0 | Mild | Severe | ... | Dementia | 81.0 | 22.3 | 22.0 | 22.3 | NaN | NaN | 3/3 | N | 0.752803 |
| H21.33.047 | Intermediate | Thal 2 | Braak V | Frequent | Not identified | Neocortical (Diffuse) | 1 | 1 | Moderate | Moderate | ... | No dementia | 90.0 | 7.4 | 26.0 | 7.4 | NaN | NaN | 3/3 | N | 0.259830 |
84 rows × 21 columns
We combine the donor, library, and disease tables into an extended cell metadata table.
cell_extended = cell.join(donor, on='donor_label')
cell_extended = cell_extended.join(library, on='library_label', rsuffix='_library_table')
cell_extended = cell_extended.join(disease, on='donor_label', rsuffix='_disease')
del cell
We use the groupby function to show the number of cells for one of disease data columns. Change this to any other column to get counts by any of the various metadata.
cell_extended.groupby('region_of_interest_name')[['region_of_interest_label']].count()
| region_of_interest_label | |
|---|---|
| region_of_interest_name | |
| angular gyrus | 389804 |
| dorsolateral prefrontal cortex | 1807513 |
| frontal agranular insular cortex (area FI) | 321843 |
| hippocampus (hippocampal formation) | 307789 |
| inferior temporal gyrus | 626792 |
| lateral (anterior) entorhinal cortex | 239721 |
| medial (posterior) entorhinal cortex | 1485051 |
| middle temporal gyrus | 1861334 |
| primary visual cortex (striate cortex, area V1/17) | 867151 |
| superior temporal gyrus | 734344 |
We can use the group by functionality to group the cells by cognitive status.
cell_extended.groupby('Cognitive Status')[['library_label']].count().rename(columns={'library_label': 'number_of_cells'})
| number_of_cells | |
|---|---|
| Cognitive Status | |
| Dementia | 3943893 |
| No dementia | 4697449 |
Adding color and feature order#
Each major feature in the donor and library table is associated with unique colors and an ordering with the set of values. Below we load the value_sets DataFrame which is a mapping from the various value in the donor and species tables to those colors and orderings. We incorporate these values into the cell metadata table.
value_sets = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-10X',
file_name='value_sets'
).set_index('label')
value_sets
value_sets.csv: 100%|██████████| 15.6k/15.6k [00:00<00:00, 111kMB/s]
| field | table | color_hex_triplet | order | description | external_identifier | |
|---|---|---|---|---|---|---|
| label | ||||||
| 2/2 | APOE Genotype | disease | #fdd4c2 | 1 | NaN | NaN |
| 2/3 | APOE Genotype | disease | #fca082 | 2 | NaN | NaN |
| 2/4 | APOE Genotype | disease | #fb6a4a | 3 | NaN | NaN |
| 3/3 | APOE Genotype | disease | #e32f27 | 4 | NaN | NaN |
| 3/4 | APOE Genotype | disease | #b21218 | 5 | NaN | NaN |
| ... | ... | ... | ... | ... | ... | ... |
| FI | region_of_interest_label | library | #008080 | 7 | agranular frontal insular cortex (area FI) | DHBA:10329 |
| STG | region_of_interest_label | library | #006400 | 8 | superior temporal gyrus | DHBA:12140 |
| DFC | region_of_interest_label | library | #00008b | 9 | dorsolateral prefrontal cortex | DHBA:10173 |
| AnG | region_of_interest_label | library | #00bfff | 10 | angular gyrus | DHBA:12136 |
| V1C | region_of_interest_label | library | #9932cc | 11 | primary visual cortex (first visual cortex, st... | DHBA:10269 |
204 rows × 6 columns
We define a convenience function to add colors for the various values in the data (e.g. unique region of interest or donor sex values).
def extract_value_set(
cell_metadata_df: pd.DataFrame,
input_value_set: pd.DataFrame,
input_value_set_label: str,
dataframe_column: Optional[str] = None
):
"""Add color and order columns to the cell metadata dataframe based on the input
value set.
Columns are added as {input_value_set_label}_color and {input_value_set_label}_order.
Parameters
----------
cell_metadata_df : pd.DataFrame
DataFrame containing cell metadata.
input_value_set : pd.DataFrame
DataFrame containing the value set information.
input_value_set_label : str
The the column name to extract color and order information for. will be added to the cell metadata.
"""
if dataframe_column is None:
dataframe_column = input_value_set_label
cell_metadata_df[f'{dataframe_column}_color'] = input_value_set[
input_value_set['field'] == input_value_set_label
].loc[cell_metadata_df[dataframe_column]]['color_hex_triplet'].values
cell_metadata_df[f'{dataframe_column}_order'] = input_value_set[
input_value_set['field'] == input_value_set_label
].loc[cell_metadata_df[dataframe_column]]['order'].values
Use our function to add the relevant color and order columns to our cell_metadata table.
# Add region of interest color and order
extract_value_set(cell_extended, value_sets, 'region_of_interest_label')
# Add disease color and order
extract_value_set(cell_extended, value_sets, 'APOE Genotype')
extract_value_set(cell_extended, value_sets, 'Arteriolosclerosis')
extract_value_set(cell_extended, value_sets, 'Atherosclerosis')
extract_value_set(cell_extended, value_sets, 'Braak')
extract_value_set(cell_extended, value_sets, 'CERAD score')
extract_value_set(cell_extended, value_sets, 'Cognitive Status')
extract_value_set(cell_extended, value_sets, 'LATE')
extract_value_set(cell_extended, value_sets, 'Highest Lewy Body Disease')
extract_value_set(cell_extended, value_sets, 'Overall AD neuropathological Change')
extract_value_set(cell_extended, value_sets, 'Severely Affected Donor')
extract_value_set(cell_extended, value_sets, 'Thal')
# Add donor sex/gender color and order
extract_value_set(cell_extended, value_sets, 'donor_sex')
extract_value_set(cell_extended, value_sets, 'donor_gender')
# Add race
extract_value_set(cell_extended, value_sets, 'donor_race')
cell_extended.head()
| cell_barcode | barcoded_cell_sample_label | library_label | alignment_job_id | doublet_score | umi_count | donor_label | exp_component_name | feature_matrix_label | dataset_label | ... | Severely Affected Donor_color | Severely Affected Donor_order | Thal_color | Thal_order | donor_sex_color | donor_sex_order | donor_gender_color | donor_gender_order | donor_race_color | donor_race_order | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_label | |||||||||||||||||||||
| AAACAGCCACTGGCTG-2001_A08 | AAACAGCCACTGGCTG | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.184615 | 54020.0 | H21.33.001 | AAACAGCCACTGGCTG-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X | ... | #fb6a4a | 1 | #fc8161 | 3 | #ADC4C3 | 2 | #ADC4C3 | 2 | #f7f184 | 125 |
| AAACAGCCAGGTTATT-2001_A08 | AAACAGCCAGGTTATT | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.060606 | 4438.0 | H21.33.001 | AAACAGCCAGGTTATT-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X | ... | #fb6a4a | 1 | #fc8161 | 3 | #ADC4C3 | 2 | #ADC4C3 | 2 | #f7f184 | 125 |
| AAACAGCCATTCCTCG-2001_A08 | AAACAGCCATTCCTCG | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.276923 | 66285.0 | H21.33.001 | AAACAGCCATTCCTCG-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X | ... | #fb6a4a | 1 | #fc8161 | 3 | #ADC4C3 | 2 | #ADC4C3 | 2 | #f7f184 | 125 |
| AAACATGCAATGCCTA-2001_A08 | AAACATGCAATGCCTA | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.060606 | 6019.0 | H21.33.001 | AAACATGCAATGCCTA-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X | ... | #fb6a4a | 1 | #fc8161 | 3 | #ADC4C3 | 2 | #ADC4C3 | 2 | #f7f184 | 125 |
| AAACATGCACCTCACC-2001_A08 | AAACATGCACCTCACC | 2001_A08 | L8XR_231221_02_D02 | 1322484698 | 0.181818 | 48653.0 | H21.33.001 | AAACATGCACCTCACC-L8XR_231221_02_D02-1322484698 | AnG-10X | SEA-AD-Multiregion-10X | ... | #fb6a4a | 1 | #fc8161 | 3 | #ADC4C3 | 2 | #ADC4C3 | 2 | #f7f184 | 125 |
5 rows × 94 columns
UMAP spatial embedding#
Now that we’ve merged our donor and library metadata into the main cells data, our next step is to plot these values in the Uniform Manifold Approximation and Projection (UMAP) for cells in the dataset. The UMAP is a dimension reduction technique that can be used for visualizing and exploring large-dimension datasets.
Below we load this 2-D embedding for a sub selection of our cells and merge the x-y coordinates into the extended cell metadata we are creating.
cell_2d_embedding_coordinates = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-taxonomy',
file_name='cell_2d_embedding_coordinates'
).set_index('cell_label')
cell_2d_embedding_coordinates.head()
cell_2d_embedding_coordinates.csv: 100%|██████████| 273M/273M [00:26<00:00, 10.5MMB/s]
| x | y | |
|---|---|---|
| cell_label | ||
| AAACAGCCACTGGCTG-2001_A08 | 7.009472 | 14.111731 |
| AAACAGCCAGGTTATT-2001_A08 | -1.319671 | -1.221017 |
| AAACAGCCATTCCTCG-2001_A08 | 17.003418 | 10.816355 |
| AAACATGCAATGCCTA-2001_A08 | 17.077744 | 2.317490 |
| AAACATGCACCTCACC-2001_A08 | 19.195032 | 1.132652 |
After joining the UMAPS coordinates into our full cell metadata table, we’ll subset every 10th cell the full dataset for ease of plotting.
cell_extended = cell_extended.join(cell_2d_embedding_coordinates, how='inner')
cell_extended = cell_extended.sample(frac=1) # shuffle the rows for plotting purposes
plot_cell_extended = cell_extended[::10]
del cell_2d_embedding_coordinates
We define a small helper function plot_umap to visualize the cells on the UMAP. In the examples below we will plot associated cell information colorized by donor age, sex, region of interest,etc.
def plot_umap(
xx: np.ndarray,
yy: np.ndarray,
cc: np.ndarray = None,
val: np.ndarray = None,
fig_width: float = 8,
fig_height: float = 8,
cmap: Optional[plt.Colormap] = None,
labels: np.ndarray = None,
term_orders: np.ndarray = None,
colorbar: bool = False,
sizes: np.ndarray = None,
limit_plot: bool = True,
fig: plt.Figure = None,
ax: plt.Axes = None,
) -> Tuple[plt.Figure, plt.Axes]:
"""
Plot a scatter plot of the UMAP coordinates.
Parameters
----------
xx : array-like
x-coordinates of the points to plot.
yy : array-like
y-coordinates of the points to plot.
cc : array-like, optional
colors of the points to plot. If None, the points will be colored by the values in `val`.
val : array-like, optional
values of the points to plot. If None, the points will be colored by the values in `cc`.
fig_width : float, optional
width of the figure in inches. Default is 8.
fig_height : float, optional
height of the figure in inches. Default is 8.
cmap : str, optional
colormap to use for coloring the points. If None, the points will be colored by the values in `cc`.
labels : array-like, optional
labels for the points to plot. If None, no labels will be added to the plot.
term_orders : array-like, optional
order of the labels for the legend. If None, the labels will be ordered by their appearance in `labels`.
colorbar : bool, optional
whether to add a colorbar to the plot. Default is False.
sizes : array-like, optional
sizes of the points to plot. If None, all points will have the same size.
"""
if sizes is None:
sizes = 1
if fig is None or ax is None:
fig, ax = plt.subplots()
fig.set_size_inches(fig_width, fig_height)
if cmap is not None:
scatt = ax.scatter(xx, yy, c=val, s=0.5, marker='.', cmap=cmap, alpha=sizes)
elif cc is not None:
scatt = ax.scatter(xx, yy, c=cc, s=0.5, marker='.', alpha=sizes)
if labels is not None:
from matplotlib.patches import Rectangle
unique_label_colors = (labels + ',' + cc).unique()
unique_labels = np.array([label_color.split(',')[0] for label_color in unique_label_colors])
unique_colors = np.array([label_color.split(',')[1] for label_color in unique_label_colors])
if term_orders is not None:
unique_order = term_orders.unique()
term_order = np.argsort(unique_order)
unique_labels = unique_labels[term_order]
unique_colors = unique_colors[term_order]
rects = []
for color in unique_colors:
rects.append(Rectangle((0, 0), 1, 1, fc=color))
legend = ax.legend(rects, unique_labels, loc=1)
# ax.add_artist(legend)
if colorbar:
fig.colorbar(scatt, ax=ax)
return fig, ax
Plot the various donor and library metadata available.
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended['donor_sex_color'],
labels=plot_cell_extended['donor_sex'],
term_orders=plot_cell_extended['donor_sex_order'],
fig_width=12,
fig_height=12
)
res = ax.set_title("donor_sex")
plt.show()
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended['donor_gender_color'],
labels=plot_cell_extended['donor_gender'],
term_orders=plot_cell_extended['donor_gender_order'],
fig_width=12,
fig_height=12
)
res = ax.set_title("donor_gender")
plt.show()
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
val=plot_cell_extended['donor_age_value'],
cmap=plt.cm.Blues,
fig_width=14,
fig_height=12,
colorbar=True,
)
res = ax.set_title("donor_age")
plt.show()
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
val=plot_cell_extended['Years of education'],
cmap=plt.cm.Blues,
fig_width=14,
fig_height=12,
colorbar=True,
)
res = ax.set_title("Years of education")
plt.show()
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended['donor_race_color'],
labels=plot_cell_extended['donor_race'],
term_orders=plot_cell_extended['donor_race_order'],
fig_width=12,
fig_height=12
)
res = ax.set_title("donor_race")
plt.show()
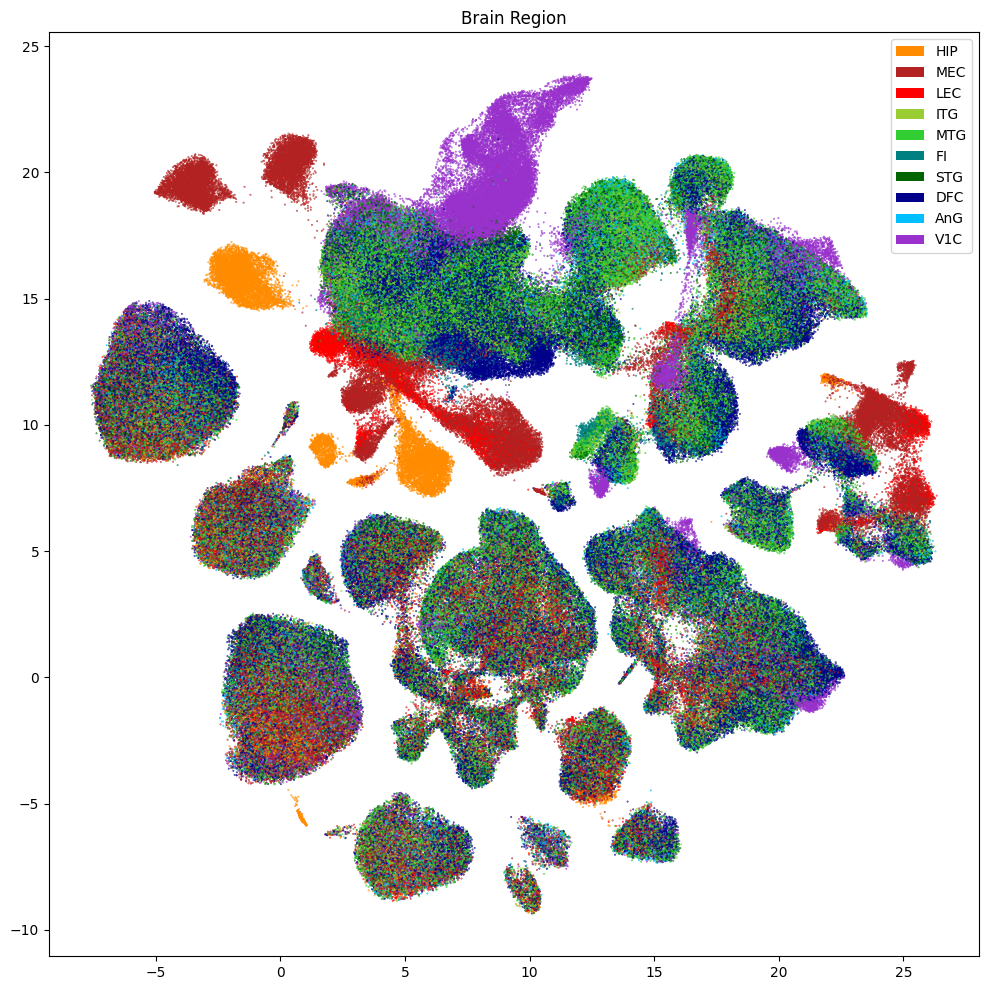
Below we show the region of interest for the cells in the dataset.
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended['region_of_interest_label_color'],
labels=plot_cell_extended['region_of_interest_label'],
term_orders=plot_cell_extended['region_of_interest_label_order'],
fig_width=12,
fig_height=12
)
res = ax.set_title("Brain Region")
plt.show()
New we plot the UMAP with for the various disease markers in the data.
for disease_value in value_sets[value_sets['table'] == 'disease']['field'].unique():
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended[f'{disease_value}_color'],
labels=plot_cell_extended[f'{disease_value}'],
term_orders=plot_cell_extended[f'{disease_value}_order'],
fig_width=12,
fig_height=12
)
res = ax.set_title(f"{disease_value}")
plt.show()
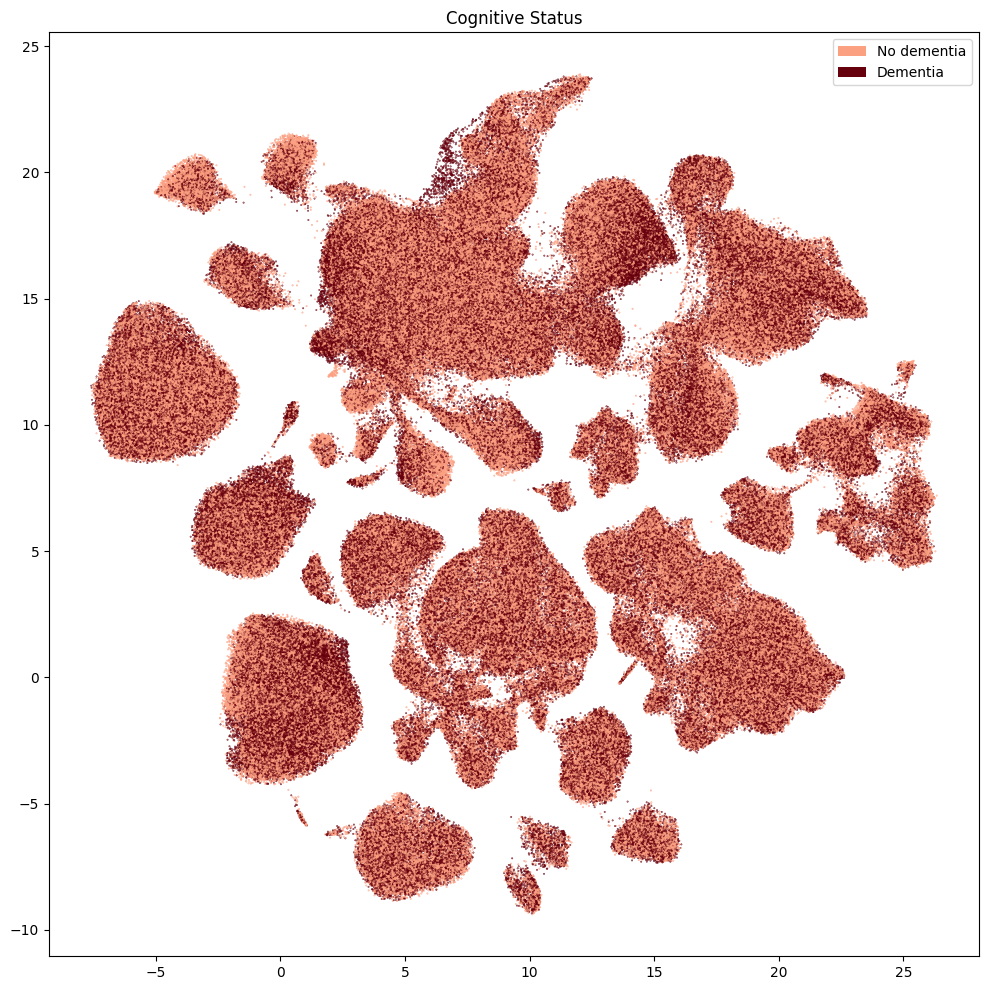
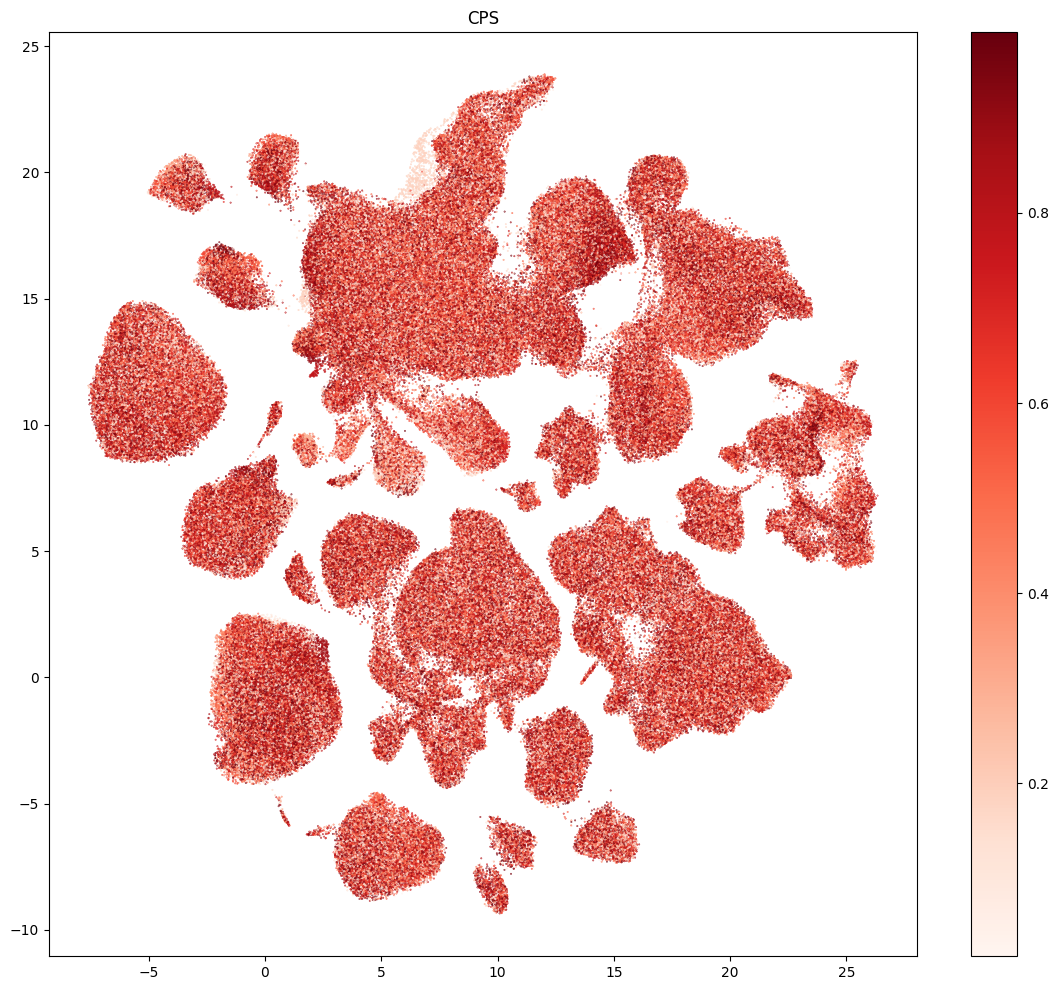
Finally, we’ll plot the pseudo-progression scores (CPS) for these data.
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
val=plot_cell_extended['CPS_Global'],
cmap=plt.cm.Reds,
fig_width=14,
fig_height=12,
colorbar=True,
)
res = ax.set_title("CPS")
plt.show()
Taxonomy Information#
The final set of metadata we load into our extended cell metadata file maps the cells into their assigned cluster in the taxonomy. We additionally load metadata for the clusters and compute useful information, such as the number of cells in each taxon at each level of the taxonomy. In this notebook cluster refers to the leaf node of the taxonomy as is the case throughout abc_atlas_access. In this taxonomy, the highest granularity cell type is called ‘supertype’; therefore, when you see the term ‘cluster’ below it refers to the leaf node of the taxonomy aka, the supertypes.
First, we load information associated with the lowest level in the taxonomy. This includes a useful alias value for each cluster as well as the number of cells in each supertype.
cluster = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-taxonomy',
file_name='cluster',
dtype={'number_of_cells': 'Int64'}
).rename(columns={'label': 'cluster_annotation_term_label'}).set_index('cluster_annotation_term_label')
cluster.head()
cluster.csv: 100%|██████████| 6.03k/6.03k [00:00<00:00, 72.4kMB/s]
| cluster_alias | number_of_cells | |
|---|---|---|
| cluster_annotation_term_label | ||
| CS20260630_SUPR_001 | 1 | 88293 |
| CS20260630_SUPR_002 | 2 | 3295 |
| CS20260630_SUPR_003 | 3 | 2368 |
| CS20260630_SUPR_004 | 4 | 700 |
| CS20260630_SUPR_005 | 5 | 14450 |
Next, we load the table that describes the levels in the taxonomy from class at the highest to supertype at the lowest level.
cluster_annotation_term_set = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-taxonomy',
file_name='cluster_annotation_term_set'
).rename(columns={'label': 'cluster_annotation_term_label'})
cluster_annotation_term_set
cluster_annotation_term_set.csv: 100%|██████████| 208/208 [00:00<00:00, 2.25kMB/s]
| name | cluster_annotation_term_label | description | order | parent_term_set_label | |
|---|---|---|---|---|---|
| 0 | Class | CCN20260630_LEVEL_0 | Class | 0 | NaN |
| 1 | Subclass | CCN20260630_LEVEL_1 | Subclass | 1 | CCN20260630_LEVEL_0 |
| 2 | Supertype | CCN20260630_LEVEL_2 | Supertype | 2 | CCN20260630_LEVEL_1 |
For the supertypes, we load information on the annotations for each supertype. This also includes the term order and color information which we will use to plot later.
cluster_annotation_term = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-taxonomy',
file_name='cluster_annotation_term',
).rename(columns={'label': 'cluster_annotation_term_label'}).set_index('cluster_annotation_term_label')
cluster_annotation_term
cluster_annotation_term.csv: 100%|██████████| 32.3k/32.3k [00:00<00:00, 241kMB/s]
| name | cluster_annotation_term_set_label | cluster_annotation_term_set_name | color_hex_triplet | term_order | term_set_order | parent_term_label | parent_term_name | parent_term_set_label | CCN20230508_label | |
|---|---|---|---|---|---|---|---|---|---|---|
| cluster_annotation_term_label | ||||||||||
| CS20260630_CLAS_001 | Neuronal: GABAergic | CCN20260630_LEVEL_0 | Class | #F05A28 | 1 | 0 | NaN | NaN | NaN | NaN |
| CS20260630_CLAS_002 | Neuronal: Glutamatergic | CCN20260630_LEVEL_0 | Class | #00ADF8 | 2 | 0 | NaN | NaN | NaN | NaN |
| CS20260630_CLAS_003 | Non-neuronal and Non-neural | CCN20260630_LEVEL_0 | Class | #808080 | 3 | 0 | NaN | NaN | NaN | NaN |
| CS20260630_SCLA_023 | Astrocyte | CCN20260630_LEVEL_1 | Subclass | #665C47 | 23 | 1 | CS20260630_CLAS_003 | Non-neuronal and Non-neural | CCN20260630_LEVEL_0 | NaN |
| CS20260630_SCLA_021 | CA2-4 | CCN20260630_LEVEL_1 | Subclass | #D5BF41 | 21 | 1 | CS20260630_CLAS_002 | Neuronal: Glutamatergic | CCN20260630_LEVEL_0 | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| CS20260630_SUPR_038 | Vip_25-SEAAD | CCN20260630_LEVEL_2 | Supertype | #492C56 | 38 | 2 | CS20260630_SCLA_005 | Vip | CCN20260630_LEVEL_1 | NaN |
| CS20260630_SUPR_039 | Vip_4 | CCN20260630_LEVEL_2 | Supertype | #D0AEDC | 39 | 2 | CS20260630_SCLA_005 | Vip | CCN20260630_LEVEL_1 | CS20230508_SUPT_0022 |
| CS20260630_SUPR_040 | Vip_5 | CCN20260630_LEVEL_2 | Supertype | #C7A5D3 | 40 | 2 | CS20260630_SCLA_005 | Vip | CCN20260630_LEVEL_1 | CS20230508_SUPT_0023 |
| CS20260630_SUPR_041 | Vip_6 | CCN20260630_LEVEL_2 | Supertype | #BE9DCA | 41 | 2 | CS20260630_SCLA_005 | Vip | CCN20260630_LEVEL_1 | CS20230508_SUPT_0024 |
| CS20260630_SUPR_042 | Vip_9 | CCN20260630_LEVEL_2 | Supertype | #B494C1 | 42 | 2 | CS20260630_SCLA_005 | Vip | CCN20260630_LEVEL_1 | CS20230508_SUPT_0025 |
239 rows × 10 columns
Finally, we load the cluster to cluster annotation membership table. Each row in this table is a mapping between the supertypes and every level of the taxonomy it belongs to, including itself. We’ll use this table in a groupby to allow us to count up the number of clusters at each taxonomy level and sum the number of cells in each taxon in the taxonomy a all levels.
cluster_to_cluster_annotation_membership = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-taxonomy',
file_name='cluster_to_cluster_annotation_membership'
).set_index('cluster_annotation_term_label')
membership_with_cluster_info = cluster_to_cluster_annotation_membership.join(
cluster.reset_index().set_index('cluster_alias')[['number_of_cells']],
on='cluster_alias'
)
membership_with_cluster_info = membership_with_cluster_info.join(cluster_annotation_term, rsuffix='_anno_term').reset_index()
membership_groupby = membership_with_cluster_info.groupby(
['cluster_alias', 'cluster_annotation_term_set_name']
)
membership_with_cluster_info.head()
cluster_to_cluster_annotation_membership.csv: 100%|██████████| 40.6k/40.6k [00:00<00:00, 334kMB/s]
| cluster_annotation_term_label | cluster_annotation_term_set_name | cluster_annotation_term_name | cluster_alias | cluster_annotation_term_set_label | number_of_cells | name | cluster_annotation_term_set_label_anno_term | cluster_annotation_term_set_name_anno_term | color_hex_triplet | term_order | term_set_order | parent_term_label | parent_term_name | parent_term_set_label | CCN20230508_label | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | CS20260630_CLAS_001 | Class | Neuronal: GABAergic | 1 | CCN20260630_LEVEL_0 | 88293 | Neuronal: GABAergic | CCN20260630_LEVEL_0 | Class | #F05A28 | 1 | 0 | NaN | NaN | NaN | NaN |
| 1 | CS20260630_CLAS_001 | Class | Neuronal: GABAergic | 2 | CCN20260630_LEVEL_0 | 3295 | Neuronal: GABAergic | CCN20260630_LEVEL_0 | Class | #F05A28 | 1 | 0 | NaN | NaN | NaN | NaN |
| 2 | CS20260630_CLAS_001 | Class | Neuronal: GABAergic | 3 | CCN20260630_LEVEL_0 | 2368 | Neuronal: GABAergic | CCN20260630_LEVEL_0 | Class | #F05A28 | 1 | 0 | NaN | NaN | NaN | NaN |
| 3 | CS20260630_CLAS_001 | Class | Neuronal: GABAergic | 4 | CCN20260630_LEVEL_0 | 700 | Neuronal: GABAergic | CCN20260630_LEVEL_0 | Class | #F05A28 | 1 | 0 | NaN | NaN | NaN | NaN |
| 4 | CS20260630_CLAS_001 | Class | Neuronal: GABAergic | 5 | CCN20260630_LEVEL_0 | 14450 | Neuronal: GABAergic | CCN20260630_LEVEL_0 | Class | #F05A28 | 1 | 0 | NaN | NaN | NaN | NaN |
From the membership table, we create three tables via a groupby. First the name of each cluster and its parents.
# term_sets = abc_cache.get_metadata_dataframe(directory='WHB-taxonomy', file_name='cluster_annotation_term_set').set_index('label')
cluster_details = membership_groupby['cluster_annotation_term_name'].first().unstack()
cluster_details = cluster_details[cluster_annotation_term_set['name']] # order columns
cluster_details.fillna('Other', inplace=True)
cluster_details.head()
| cluster_annotation_term_set_name | Class | Subclass | Supertype |
|---|---|---|---|
| cluster_alias | |||
| 1 | Neuronal: GABAergic | Lamp5 Lhx6 | Lamp5_Lhx6_1 |
| 2 | Neuronal: GABAergic | Lamp5 Lhx6 | Lamp5_Lhx6_2-SEAAD |
| 3 | Neuronal: GABAergic | Lamp5 Lhx6 | Lamp5_Lhx6_3-SEAAD |
| 4 | Neuronal: GABAergic | Lamp5 Lhx6 | Lamp5_Lhx6_4-SEAAD |
| 5 | Neuronal: GABAergic | Lamp5 | Lamp5_1 |
Next the plotting order of each of the taxons and their parents.
cluster_order = membership_groupby['term_order'].first().unstack()
cluster_order.head()
| cluster_annotation_term_set_name | Class | Subclass | Supertype |
|---|---|---|---|
| cluster_alias | |||
| 1 | 1 | 1 | 1 |
| 2 | 1 | 1 | 2 |
| 3 | 1 | 1 | 3 |
| 4 | 1 | 1 | 4 |
| 5 | 1 | 2 | 5 |
Finally, the colors we will use to plot for each of the unique taxons at all levels.
cluster_colors = membership_groupby['color_hex_triplet'].first().unstack()
cluster_colors = cluster_colors[cluster_annotation_term_set['name']]
cluster_colors.head()
| cluster_annotation_term_set_name | Class | Subclass | Supertype |
|---|---|---|---|
| cluster_alias | |||
| 1 | #F05A28 | #935F50 | #C6A299 |
| 2 | #F05A28 | #935F50 | #A68880 |
| 3 | #F05A28 | #935F50 | #866E67 |
| 4 | #F05A28 | #935F50 | #67544F |
| 5 | #F05A28 | #DA808C | #F9B7C3 |
Next, we bring it all together by loading the mapping of cells to supertype and join into our final metadata table.
cell_to_cluster_membership = abc_cache.get_metadata_dataframe(
directory='SEA-AD-Multiregion-taxonomy',
file_name='cell_to_cluster_membership',
).set_index('cell_label')
cell_to_cluster_membership.head()
cell_to_cluster_membership.csv: 100%|██████████| 295M/295M [00:25<00:00, 11.8MMB/s]
| cluster_alias | label | |
|---|---|---|
| cell_label | ||
| AAACAGCCACTGGCTG-2001_A08 | 84 | CS20260630_SUPR_084 |
| AAACAGCCAGGTTATT-2001_A08 | 180 | CS20260630_SUPR_180 |
| AAACAGCCATTCCTCG-2001_A08 | 113 | CS20260630_SUPR_113 |
| AAACATGCAATGCCTA-2001_A08 | 80 | CS20260630_SUPR_080 |
| AAACATGCACCTCACC-2001_A08 | 76 | CS20260630_SUPR_076 |
We merge this table with information from our taxons. Again, we’ll subset the full set of cells for ease of plotting.
cell_extended = cell_extended.join(cell_to_cluster_membership, rsuffix='_cell_to_cluster_membership', how='inner')
cell_extended = cell_extended.join(cluster_details, on='cluster_alias')
cell_extended = cell_extended.join(cluster_colors, on='cluster_alias', rsuffix='_color')
cell_extended = cell_extended.join(cluster_order, on='cluster_alias', rsuffix='_order')
plot_cell_extended = cell_extended[::10]
del cell_to_cluster_membership
cell_extended.head()
| cell_barcode | barcoded_cell_sample_label | library_label | alignment_job_id | doublet_score | umi_count | donor_label | exp_component_name | feature_matrix_label | dataset_label | ... | label | Class | Subclass | Supertype | Class_color | Subclass_color | Supertype_color | Class_order | Subclass_order | Supertype_order | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_label | |||||||||||||||||||||
| CAATTTCAGCCTTTGA-1580_C01 | CAATTTCAGCCTTTGA | 1580_C01 | L8HX_230126_02_E07 | 1244668541 | 0.100000 | 24846.0 | H20.33.040 | CAATTTCAGCCTTTGA-L8HX_230126_02_E07-1244668541 | STG-10X | SEA-AD-Multiregion-10X | ... | CS20260630_SUPR_025 | Neuronal: GABAergic | Vip | Vip_1 | #F05A28 | #A45FBF | #E2C0EE | 1 | 5 | 25 |
| AGGTGTTGTGGTTTGT-633_F06 | AGGTGTTGTGGTTTGT | 633_F06 | L8TX_210506_01_E08 | 1153814230 | 0.060000 | 7716.0 | H21.33.022 | AGGTGTTGTGGTTTGT-L8TX_210506_01_E08-1153814230 | MTG-10X | SEA-AD-Multiregion-10X | ... | CS20260630_SUPR_036 | Neuronal: GABAergic | Vip | Vip_23 | #F05A28 | #A45FBF | #5A3D67 | 1 | 5 | 36 |
| AATGCCAGTTCAAGGG-1725_C01 | AATGCCAGTTCAAGGG | 1725_C01 | L8HX_230601_22_D04 | 1277848644 | 0.060000 | 32147.0 | H19.33.004 | AATGCCAGTTCAAGGG-L8HX_230601_22_D04-1277848644 | ITG-10X | SEA-AD-Multiregion-10X | ... | CS20260630_SUPR_092 | Neuronal: Glutamatergic | L2/3 IT | L2/3 IT_6 | #00ADF8 | #B1EC30 | #B0BF64 | 2 | 10 | 92 |
| TACCTCGGTCAAAGAT-733_G01 | TACCTCGGTCAAAGAT | 733_G01 | L8TX_210722_01_B08 | 1153814305 | 0.020833 | 10344.0 | H21.33.033 | TACCTCGGTCAAAGAT-L8TX_210722_01_B08-1153814305 | MTG-10X | SEA-AD-Multiregion-10X | ... | CS20260630_SUPR_084 | Neuronal: Glutamatergic | L2/3 IT | L2/3 IT_1 | #00ADF8 | #B1EC30 | #EEF987 | 2 | 10 | 84 |
| GCACGGTTCTTCGTGC-763_H06 | GCACGGTTCTTCGTGC | 763_H06 | L8TX_210805_01_E02 | 1124416548 | 0.312500 | 59494.0 | H20.33.020 | GCACGGTTCTTCGTGC-L8TX_210805_01_E02-1124416548 | DFC-10X | SEA-AD-Multiregion-10X | ... | CS20260630_SUPR_041 | Neuronal: GABAergic | Vip | Vip_6 | #F05A28 | #A45FBF | #BE9DCA | 1 | 5 | 41 |
5 rows × 107 columns
print_column_info(cell_extended)
Number of unique cell_barcode = 3264931
Number of unique barcoded_cell_sample_label = 907
Number of unique library_label = 907
Number of unique alignment_job_id = 912
Number of unique doublet_score = 2395
Number of unique umi_count = 164013
Number of unique donor_label = 84
Number of unique exp_component_name = 6013346
Number of unique feature_matrix_label = 10 ['AnG-10X', 'DFC-10X', 'FI-10X', 'HIP-10X', 'ITG-10X', 'LEC-10X', 'MEC-10X', 'MTG-10X', 'STG-10X', 'V1C-10X']
Number of unique dataset_label = 1 ['SEA-AD-Multiregion-10X']
Number of unique donor_species = 1 ['NCBITaxon:9606']
Number of unique species_scientific_name = 1 ['Homo sapiens']
Number of unique species_genus = 1 ['Human']
Number of unique donor_sex = 2 ['Female', 'Male']
Number of unique donor_gender = 2 ['Female', 'Male']
Number of unique donor_age = 29 ['100+ yrs', '65 yrs', '68 yrs', '69 yrs', '70 yrs', '72 yrs', '75 yrs', '77 yrs', '78 yrs', '80 yrs', '81 yrs', '82 yrs', '83 yrs', '84 yrs', '85 yrs', '86 yrs', '87 yrs', '88 yrs', '89 yrs', '90 yrs', '91 yrs', '92 yrs', '93 yrs', '94 yrs', '95 yrs', '96 yrs', '97 yrs', '98 yrs', '99 yrs']
Number of unique donor_age_value = 29 [65, 68, 69, 70, 72, 75, 77, 78, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100]
Number of unique donor_age_unit = 2 ['+ yrs_not_in_mapping', 'years']
Number of unique Race (choice=White) = 2 ['Checked', 'Unchecked']
Number of unique Race (choice=Black/ African American) = 1 ['Unchecked']
Number of unique Race (choice=Asian) = 2 ['Checked', 'Unchecked']
Number of unique Race (choice=American Indian/ Alaska Native) = 2 ['Checked', 'Unchecked']
Number of unique Race (choice=Native Hawaiian or Pacific Islander) = 1 ['Unchecked']
Number of unique Race (choice=Unknown or unreported) = 1 ['Unchecked']
Number of unique Race (choice=Other) = 2 ['Checked', 'Unchecked']
Number of unique Hispanic/Latino = 3 ['No', 'Unknown', 'Yes']
Number of unique specify other race = 1 ['Mixed']
Number of unique Highest level of education = 5 ['Bachelors', 'Graduate (PhD/Masters)', 'High School', 'Professional', 'Trade School/ Tech School']
Number of unique Years of education = 10 [12, 13, 14, 15, 16, 17, 18, 19, 20, 21]
Number of unique PMI = 60
Number of unique Fresh Brain Weight = 74
Number of unique Brain pH = 14 [4.5, 6.0, 6.2, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, 7.0, 7.2, 7.3, 7.4, 7.6]
Number of unique Primary Study Name = 2 ['ACT', 'ADRC Clinical Core']
Number of unique donor_race = 4 ['American Indian/Alaska Native/White/Other', 'Asian', 'White', 'White/Other']
Number of unique library_method = 3 ['10xMultiome;GEX', '10xV3.1', '10xV3.1_HT']
Number of unique barcoded_cell_sample_label_library_table = 907
Number of unique enrichment_population = 21 ['100% NeuN+', '38% NeuN+, 62% NeuN-', '40% NeuN+, 60% NeuN-', '44% NeuN+, 56% NeuN-', '46% NeuN+, 54% NeuN-', '50% NeuN+, 50% NeuN-', '56% NeuN+, 44% NeuN-', '57% NeuN+, 43% NeuN-', '60% NeuN+, 40% NeuN-', '62% NeuN+, 38% NeuN-', '63% NeuN+, 37% NeuN-', '64% NeuN+, 36% NeuN-', '65% NeuN+, 35% NeuN-', '66% NeuN+, 34% NeuN-', '68% NeuN+, 32% NeuN-', '70% NeuN +, 30% NeuN-', '70% NeuN+, 30% NeuN-', '71% NeuN+, 29% NeuN-', '74% NeuN+, 26% NeuN-', '90% NeuN+, 10% NeuN-', 'NeuN-positive and NeuN-negative']
Number of unique cell_specimen_type = 1 ['Nuclei']
Number of unique region_of_interest_label = 10 ['AnG', 'DFC', 'FI', 'HIP', 'ITG', 'LEC', 'MEC', 'MTG', 'STG', 'V1C']
Number of unique region_of_interest_name = 10 ['angular gyrus', 'dorsolateral prefrontal cortex', 'frontal agranular insular cortex (area FI)', 'hippocampus (hippocampal formation)', 'inferior temporal gyrus', 'lateral (anterior) entorhinal cortex', 'medial (posterior) entorhinal cortex', 'middle temporal gyrus', 'primary visual cortex (striate cortex, area V1/17)', 'superior temporal gyrus']
Number of unique parcellation_term_identifier = 10 ['DHBA:10173', 'DHBA:10269', 'DHBA:10294', 'DHBA:10318', 'DHBA:10319', 'DHBA:10329', 'DHBA:12136', 'DHBA:12140', 'DHBA:12141', 'DHBA:12142']
Number of unique Brain Region = 10 ['AnG', 'DFC', 'FI', 'HIP', 'ITG', 'LEC', 'MEC', 'MTG', 'STG', 'V1C']
Number of unique donor_label_library_table = 84
Number of unique Overall AD neuropathological Change = 4 ['High', 'Intermediate', 'Low', 'Not AD']
Number of unique Thal = 6 ['Thal 0', 'Thal 1', 'Thal 2', 'Thal 3', 'Thal 4', 'Thal 5']
Number of unique Braak = 6 ['Braak 0', 'Braak II', 'Braak III', 'Braak IV', 'Braak V', 'Braak VI']
Number of unique CERAD score = 4 ['Absent', 'Frequent', 'Moderate', 'Sparse']
Number of unique Overall CAA Score = 4 ['Mild', 'Moderate', 'Not identified', 'Severe']
Number of unique Highest Lewy Body Disease = 7 ['Amygdala-predominant', 'Brainstem-predominant', 'Limbic (Transitional)', 'Neocortical (Diffuse)', 'Not Identified (olfactory bulb assessed)', 'Not Identified (olfactory bulb not assessed)', 'Olfactory bulb only']
Number of unique Total Microinfarcts (not observed grossly) = 10 [0, 1, 2, 3, 4, 5, 7, 9, 11, 15]
Number of unique Total microinfarcts in screening sections = 9 [0, 1, 2, 3, 4, 5, 8, 9, 10]
Number of unique Atherosclerosis = 4 ['Mild', 'Moderate', 'None/NA', 'Severe']
Number of unique Arteriolosclerosis = 3 ['Mild', 'Moderate', 'Severe']
Number of unique LATE = 5 ['LATE Stage 1', 'LATE Stage 2', 'LATE Stage 3', 'Not Identified', 'Unclassifiable']
Number of unique Cognitive Status = 2 ['Dementia', 'No dementia']
Number of unique Last CASI Score = 29 [66.0, 67.0, 68.0, 70.0, 71.0, 74.0, 75.0, 77.0, 78.0, 79.0, 80.0, 81.0, 83.0, 84.0, 85.0, 86.0, 87.0, 88.0, 89.0, 90.0, 91.0, 92.0, 93.0, 94.0, 95.0, 96.0, 97.0, 98.0, 99.0]
Number of unique Interval from last CASI in months = 60
Number of unique Last MMSE Score = 18 [6.0, 11.0, 14.0, 17.0, 18.0, 19.0, 20.0, 21.0, 22.0, 23.0, 24.0, 25.0, 26.0, 27.0, 28.0, 29.0, 30.0, 33.0]
Number of unique Interval from last MMSE in months = 70
Number of unique Last MOCA Score = 10 [4.0, 7.0, 14.0, 16.0, 17.0, 19.0, 20.0, 21.0, 23.0, 25.0]
Number of unique Interval from last MOCA in months = 14 [10.1, 11.1, 14.9, 15.2, 15.8, 22.1, 23.9, 33.4, 36.1, 39.5, 41.2, 45.1, 51.3, 62.2]
Number of unique APOE Genotype = 6 ['2/2', '2/3', '2/4', '3/3', '3/4', '4/4']
Number of unique Severely Affected Donor = 2 ['N', 'Y']
Number of unique CPS_Global = 84
Number of unique region_of_interest_label_color = 10 ['#00008b', '#006400', '#008080', '#00bfff', '#32cd32', '#9932cc', '#9acd32', '#b22222', '#ff0000', '#ff8c00']
Number of unique region_of_interest_label_order = 10 [1, 3, 4, 5, 6, 7, 8, 9, 10, 11]
Number of unique APOE Genotype_color = 6 ['#67000d', '#b21218', '#e32f27', '#fb6a4a', '#fca082', '#fdd4c2']
Number of unique APOE Genotype_order = 6 [1, 2, 3, 4, 5, 6]
Number of unique Arteriolosclerosis_color = 3 ['#3f007d', '#796eb2', '#c6c7e1']
Number of unique Arteriolosclerosis_order = 3 [1, 2, 3]
Number of unique Atherosclerosis_color = 4 ['#3f007d', '#6a51a3', '#9e9ac8', '#dadaeb']
Number of unique Atherosclerosis_order = 4 [1, 2, 3, 4]
Number of unique Braak_color = 6 ['#67000d', '#aa1016', '#d52221', '#f44f39', '#fc8161', '#fedbcb']
Number of unique Braak_order = 6 [1, 3, 4, 5, 6, 7]
Number of unique CERAD score_color = 4 ['#67000d', '#cb181d', '#fb6a4a', '#fcbba1']
Number of unique CERAD score_order = 4 [1, 2, 3, 4]
Number of unique Cognitive Status_color = 2 ['#67000d', '#fca082']
Number of unique Cognitive Status_order = 2 [1, 3]
Number of unique LATE_color = 5 ['#3f007d', '#61409b', '#8683bd', '#b6b6d8', '#e2e2ef']
Number of unique LATE_order = 5 [1, 2, 3, 4, 5]
Number of unique Highest Lewy Body Disease_color = 7 ['#3f007d', '#61409b', '#9e9ac8', '#b6b6d8', '#cecee5', '#e2e2ef', '#f2f0f7']
Number of unique Highest Lewy Body Disease_order = 7 [1, 2, 3, 4, 5, 8, 10]
Number of unique Overall AD neuropathological Change_color = 4 ['#67000d', '#cb181d', '#fb6a4a', '#fcbba1']
Number of unique Overall AD neuropathological Change_order = 4 [1, 2, 3, 4]
Number of unique Severely Affected Donor_color = 2 ['#67000d', '#fb6a4a']
Number of unique Severely Affected Donor_order = 2 [1, 2]
Number of unique Thal_color = 6 ['#67000d', '#d52221', '#f44f39', '#fc8161', '#fcaf94', '#fedbcb']
Number of unique Thal_order = 6 [1, 2, 3, 4, 5, 7]
Number of unique donor_sex_color = 2 ['#565353', '#ADC4C3']
Number of unique donor_sex_order = 2 [1, 2]
Number of unique donor_gender_color = 2 ['#565353', '#ADC4C3']
Number of unique donor_gender_order = 2 [1, 2]
Number of unique donor_race_color = 4 ['#19d79d', '#bc6b63', '#e6fd9a', '#f7f184']
Number of unique donor_race_order = 4 [64, 65, 125, 126]
Number of unique x = 5550464
Number of unique y = 5397802
Number of unique cluster_alias = 207
Number of unique label = 207
Number of unique Class = 3 ['Neuronal: GABAergic', 'Neuronal: Glutamatergic', 'Non-neuronal and Non-neural']
Number of unique Subclass = 29 ['Astrocyte', 'CA2-4', 'Chandelier', 'DG', 'EC IT', 'Endothelial', 'Ependymal', 'Immune', 'L2/3 IT', 'L4 IT', 'L5 ET', 'L5 IT', 'L5/6 NP', 'L6 CT', 'L6 IT', 'L6 IT Car3', 'L6b', 'Lamp5', 'Lamp5 Lhx6', 'OPC', 'Oligodendrocyte', 'Pax6', 'Pvalb', 'Sncg', 'Sst', 'Sst Chodl', 'Sub-CA1', 'VLMC & Perivascular', 'Vip']
Number of unique Supertype = 207
Number of unique Class_color = 3 ['#00ADF8', '#808080', '#F05A28']
Number of unique Subclass_color = 29 ['#00E5E5', '#0D5B78', '#2D8CB8', '#374A45', '#3E9E64', '#50B2AD', '#5100FF', '#53776C', '#665C47', '#697255', '#7044AA', '#71238C', '#73BC48', '#7A5151', '#8D6C62', '#935F50', '#94AF97', '#A19922', '#A45FBF', '#B1B10C', '#B1EC30', '#D5BF41', '#D8442B', '#D93137', '#DA808C', '#DECB54', '#DF70FF', '#F641A8', '#FF9900']
Number of unique Supertype_color = 207
Number of unique Class_order = 3 [1, 2, 3]
Number of unique Subclass_order = 29 [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29]
Number of unique Supertype_order = 207
Plotting the taxonomy#
Now that we have our cells with associated taxonomy information, we’ll plot them into the UMAP we showed previously.
Below we plot the taxonomy mapping of the cells for each level in the taxonomy.
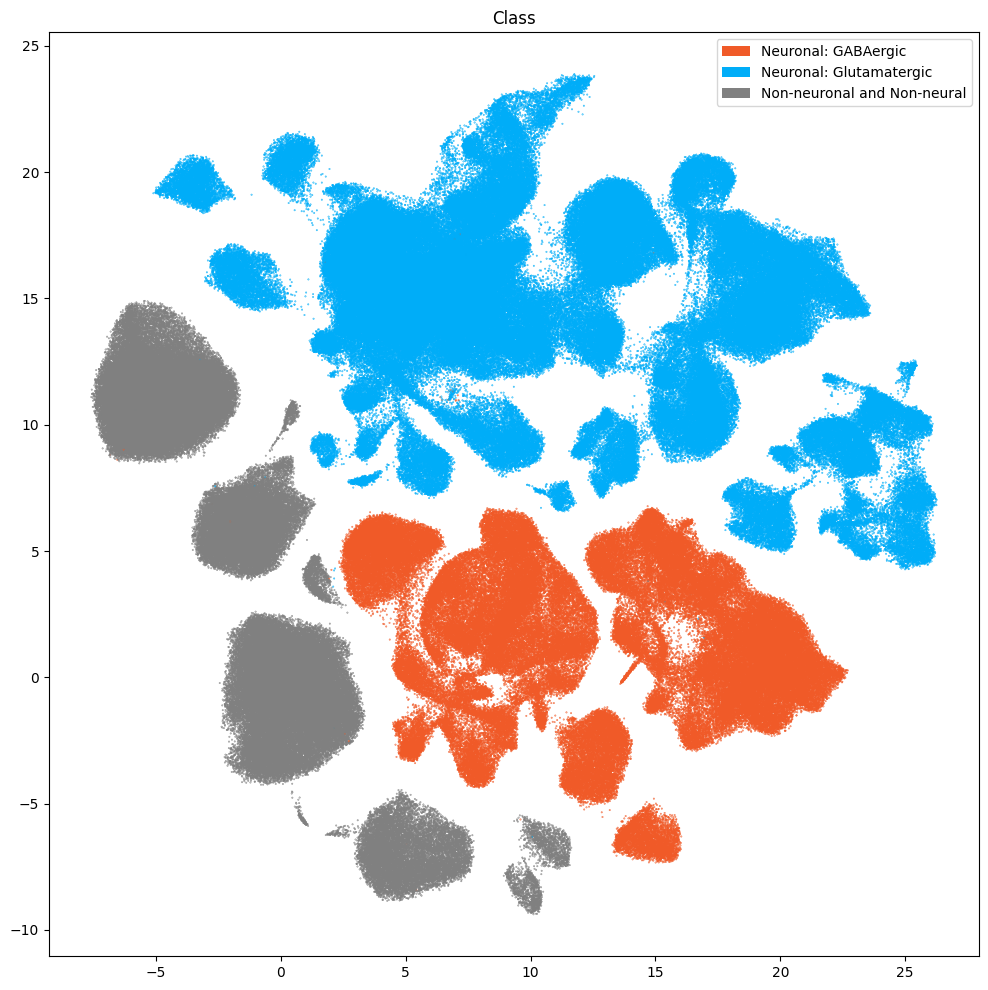
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended['Class_color'],
labels=plot_cell_extended['Class'],
term_orders=plot_cell_extended['Class_order'],
fig_width=12,
fig_height=12
)
res = ax.set_title("Class")
plt.show()
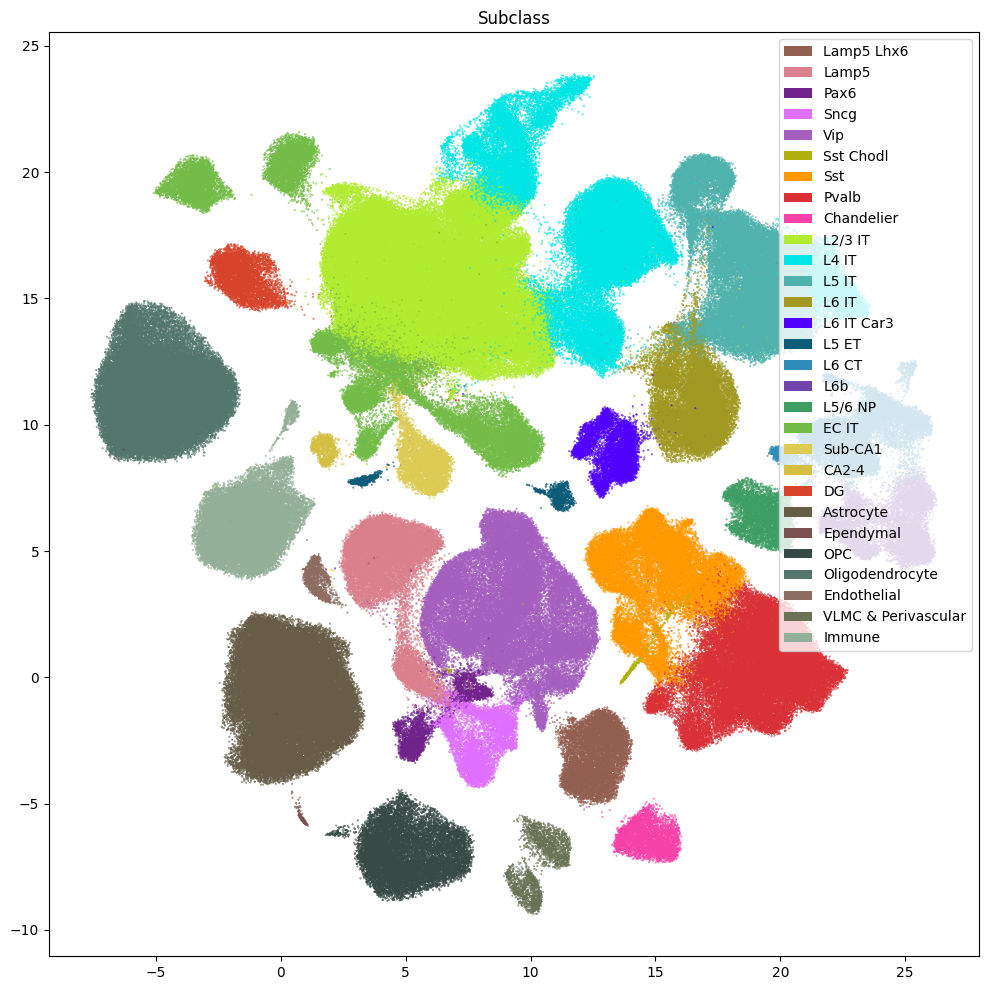
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended['Subclass_color'],
labels=plot_cell_extended['Subclass'],
term_orders=plot_cell_extended['Subclass_order'],
fig_width=12,
fig_height=12
)
res = ax.set_title("Subclass")
plt.show()
fig, ax = plot_umap(
plot_cell_extended['x'],
plot_cell_extended['y'],
cc=plot_cell_extended['Supertype_color'],
fig_width=12,
fig_height=12
)
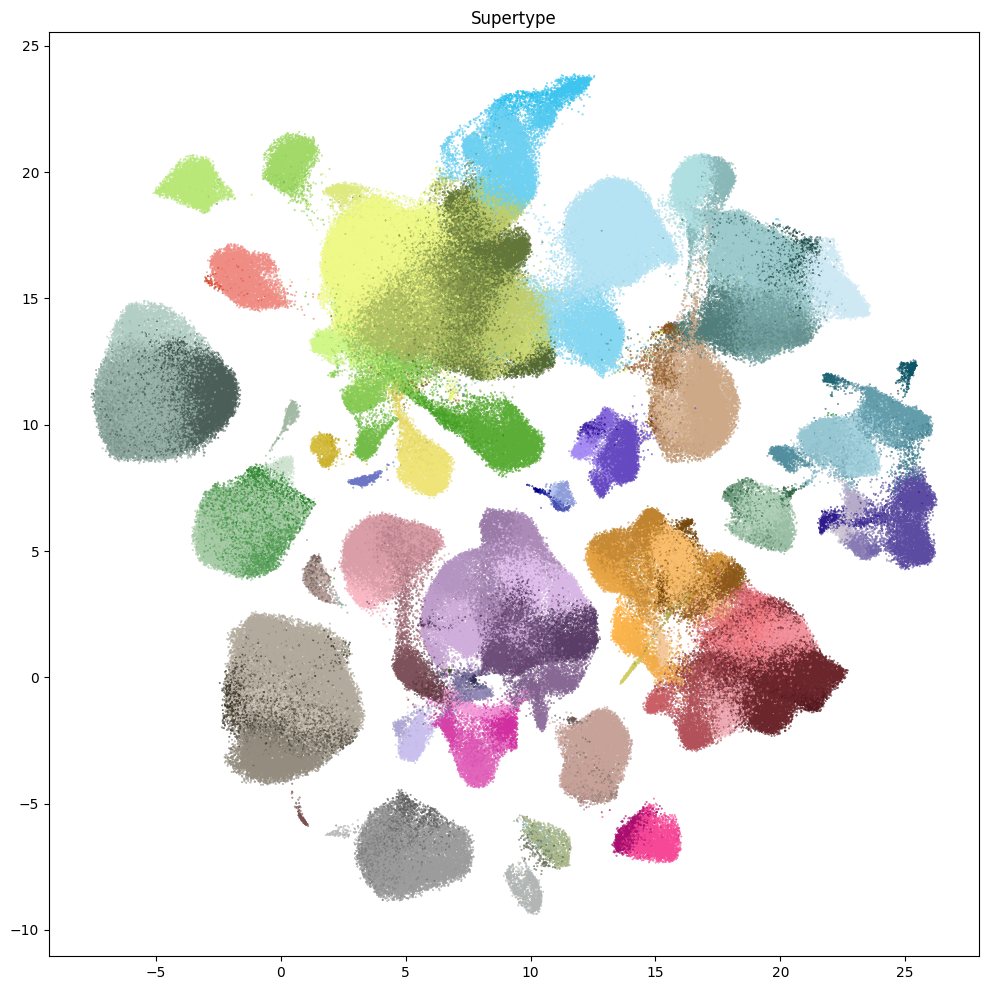
res = ax.set_title("Supertype")
plt.show()
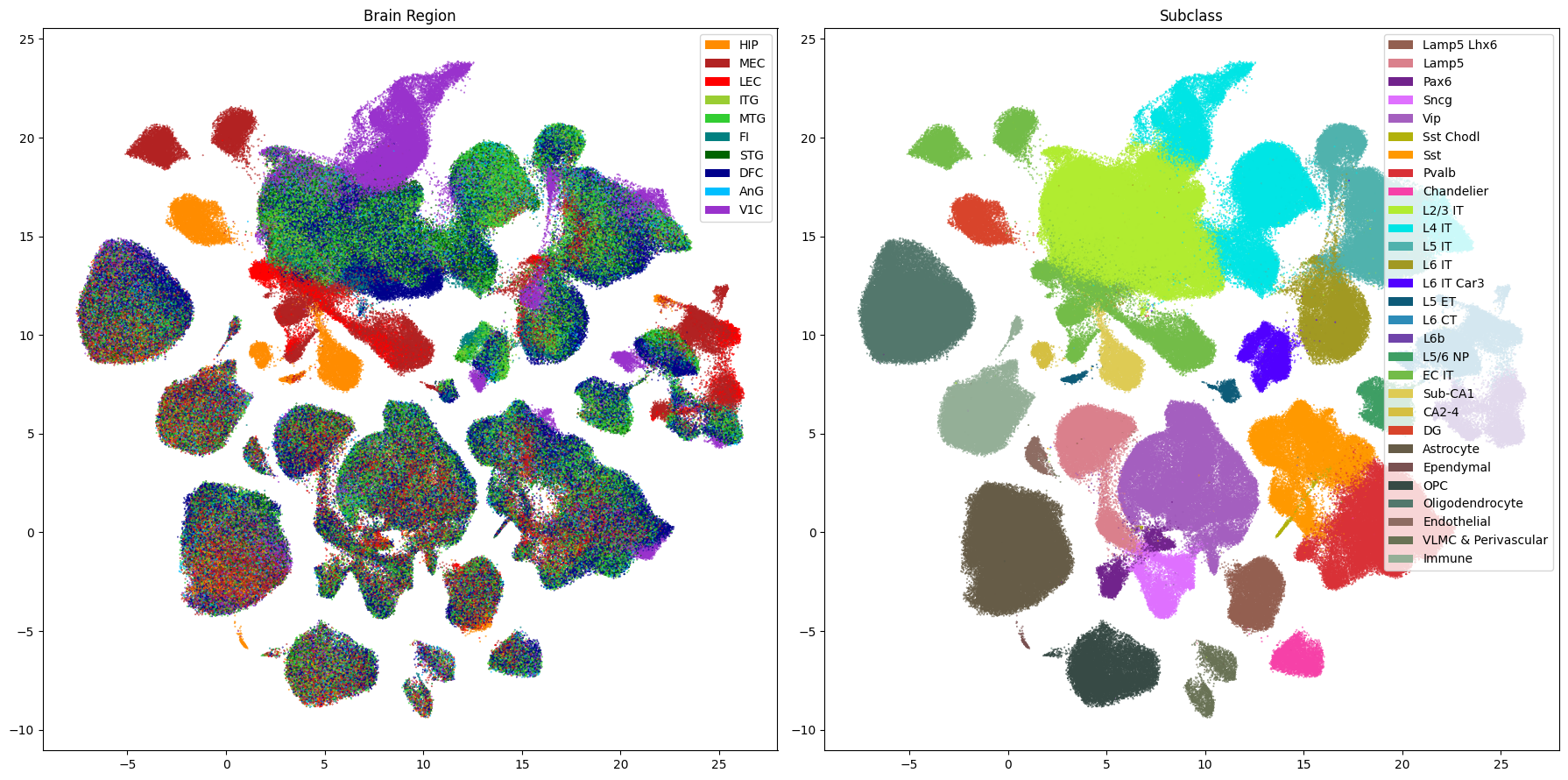
Below we plot the Subclass level of the taxonomy against paired with a plot Brain Regions in the UMAP.
fig, ax = plt.subplots(1, 2)
fig.set_size_inches(18, 9)
ax = ax.flatten()
term_to_plot_1 = 'region_of_interest_label'
plot_umap(
cell_extended['x'][::10],
cell_extended['y'][::10],
cc=cell_extended[term_to_plot_1 + '_color'][::10],
labels=cell_extended[term_to_plot_1][::10],
term_orders=cell_extended[term_to_plot_1 + '_order'][::10],
fig=fig,
ax=ax[0],
)
ax[0].set_title('Brain Region')
term_to_plot_2 = 'Subclass'
plot_umap(
cell_extended['x'][::10],
cell_extended['y'][::10],
cc=cell_extended[term_to_plot_2 + '_color'][::10],
labels=cell_extended[term_to_plot_2][::10],
term_orders=cell_extended[term_to_plot_2 + '_order'][::10],
fig=fig,
ax=ax[1],
)
ax[1].set_title(f'Subclass')
plt.tight_layout()
plt.show()
Aggregating supertype and cells counts.#
Let’s investigate the taxonomy information a bit more. In this section, we’ll create bar plots showing the number of supertypes and cells at each level in the taxonomy.
First, we need to compute the number of supertype that are in each of the cell type taxons above it. This is accomplished by a simple groupby in Pandas.
term_cluster_count = membership_with_cluster_info.reset_index().groupby(
['cluster_annotation_term_label']
)[['cluster_alias']].count()
term_cluster_count.columns = ['number_of_supertypes']
term_cluster_count.head()
| number_of_supertypes | |
|---|---|
| cluster_annotation_term_label | |
| CS20260630_CLAS_001 | 83 |
| CS20260630_CLAS_002 | 91 |
| CS20260630_CLAS_003 | 33 |
| CS20260630_SCLA_001 | 4 |
| CS20260630_SCLA_002 | 6 |
term_cell_count = membership_with_cluster_info.reset_index().groupby(
['cluster_annotation_term_label']
)[['number_of_cells']].sum()
term_cell_count.head()
| number_of_cells | |
|---|---|
| cluster_annotation_term_label | |
| CS20260630_CLAS_001 | 1421338 |
| CS20260630_CLAS_002 | 3110538 |
| CS20260630_CLAS_003 | 1481470 |
| CS20260630_SCLA_001 | 94656 |
| CS20260630_SCLA_002 | 175755 |
# Join counts with the term dataframe
term_with_counts = cluster_annotation_term.join(term_cluster_count)
term_with_counts = term_with_counts.join(term_cell_count)
term_with_counts.head()
| name | cluster_annotation_term_set_label | cluster_annotation_term_set_name | color_hex_triplet | term_order | term_set_order | parent_term_label | parent_term_name | parent_term_set_label | CCN20230508_label | number_of_supertypes | number_of_cells | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cluster_annotation_term_label | ||||||||||||
| CS20260630_CLAS_001 | Neuronal: GABAergic | CCN20260630_LEVEL_0 | Class | #F05A28 | 1 | 0 | NaN | NaN | NaN | NaN | 83 | 1421338 |
| CS20260630_CLAS_002 | Neuronal: Glutamatergic | CCN20260630_LEVEL_0 | Class | #00ADF8 | 2 | 0 | NaN | NaN | NaN | NaN | 91 | 3110538 |
| CS20260630_CLAS_003 | Non-neuronal and Non-neural | CCN20260630_LEVEL_0 | Class | #808080 | 3 | 0 | NaN | NaN | NaN | NaN | 33 | 1481470 |
| CS20260630_SCLA_023 | Astrocyte | CCN20260630_LEVEL_1 | Subclass | #665C47 | 23 | 1 | CS20260630_CLAS_003 | Non-neuronal and Non-neural | CCN20260630_LEVEL_0 | NaN | 6 | 511124 |
| CS20260630_SCLA_021 | CA2-4 | CCN20260630_LEVEL_1 | Subclass | #D5BF41 | 21 | 1 | CS20260630_CLAS_002 | Neuronal: Glutamatergic | CCN20260630_LEVEL_0 | NaN | 4 | 10572 |
Below we create a function to plot the supertype and cell counts in a bar graph, coloring by the associated taxon level.
def bar_plot_by_level_and_type(
df: pd.DataFrame,
level: str, fig_width:
float = 8.5, fig_height:
float = 4,
cell_min: int = None,
supertype_min: int = None
):
"""Plot the number of cells by the specified level.
Parameters
----------
df : pd.DataFrame
DataFrame containing cluster annotation terms with counts.
level : str
The level of the taxonomy to plot (e.g., 'Neighborhood', 'Class', 'Subclass', 'Group', 'Cluster').
fig_width : float, optional
Width of the figure in inches. Default is 8.5.
fig_height : float, optional
Height of the figure in inches. Default is 4.
"""
fig, ax = plt.subplots(1, 2)
fig.set_size_inches(fig_width, fig_height)
for idx, ctype in enumerate(['supertypes', 'cells']):
pred = (df['cluster_annotation_term_set_name'] == level)
sort_order = np.argsort(df[pred]['term_order'])
names = df[pred]['name'].iloc[sort_order]
counts = df[pred]['number_of_%s' % ctype].iloc[sort_order]
colors = df[pred]['color_hex_triplet'].iloc[sort_order]
ax[idx].barh(names, counts, color=colors)
ax[idx].set_title('Number of %s by %s' % (ctype,level))
ax[idx].set_xlabel('Number of %s' % ctype)
if ctype == 'supertypes' and supertype_min is not None:
ax[idx].set_xlim(left=supertype_min)
if ctype == 'cells':
if cell_min is not None:
ax[idx].set_xlim(left=cell_min)
ax[idx].set_xscale('log')
if idx > 0:
ax[idx].set_yticklabels([])
return fig, ax
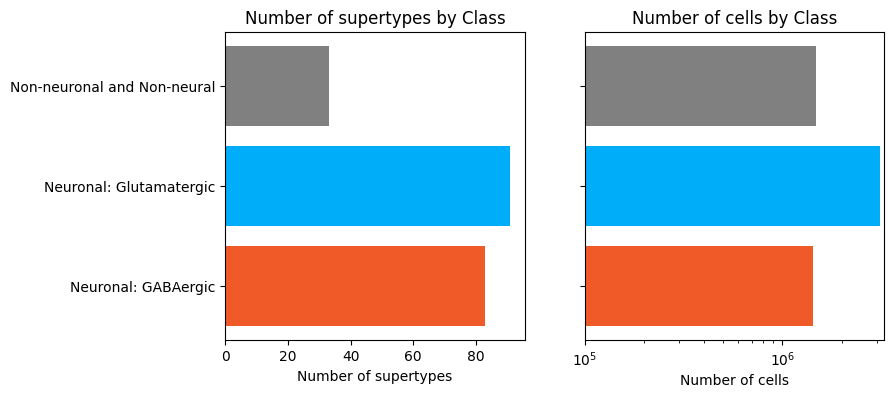
Now let’s plot the counts the taxonomy levels class and subclass.
fig, ax = bar_plot_by_level_and_type(term_with_counts, 'Class', cell_min=10 ** 5)
plt.show()
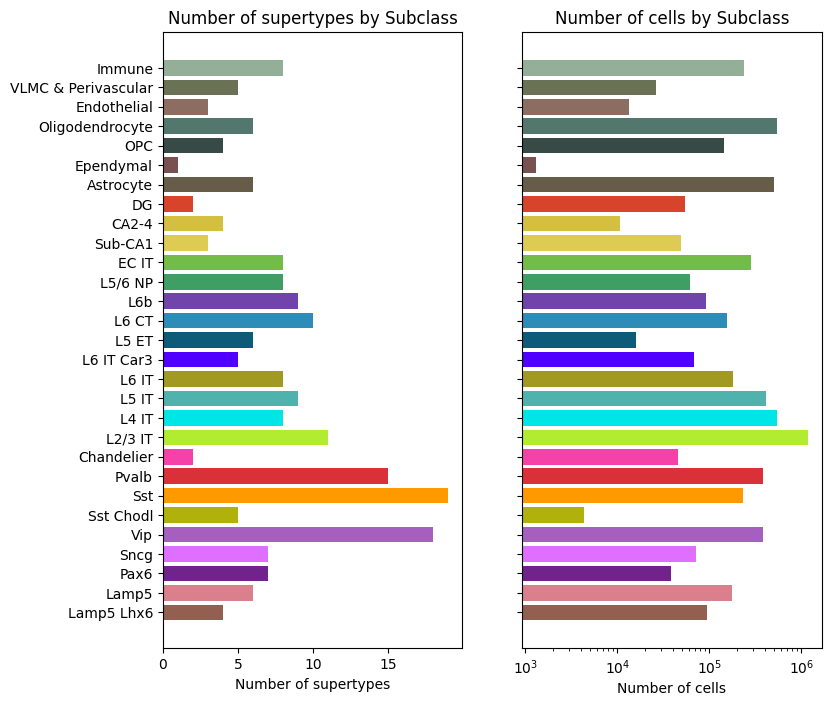
fig, ax = bar_plot_by_level_and_type(
term_with_counts,
'Subclass',
fig_height=8
)
plt.show()
Visualizing the SEA-AD Multiregion Taxonomy#
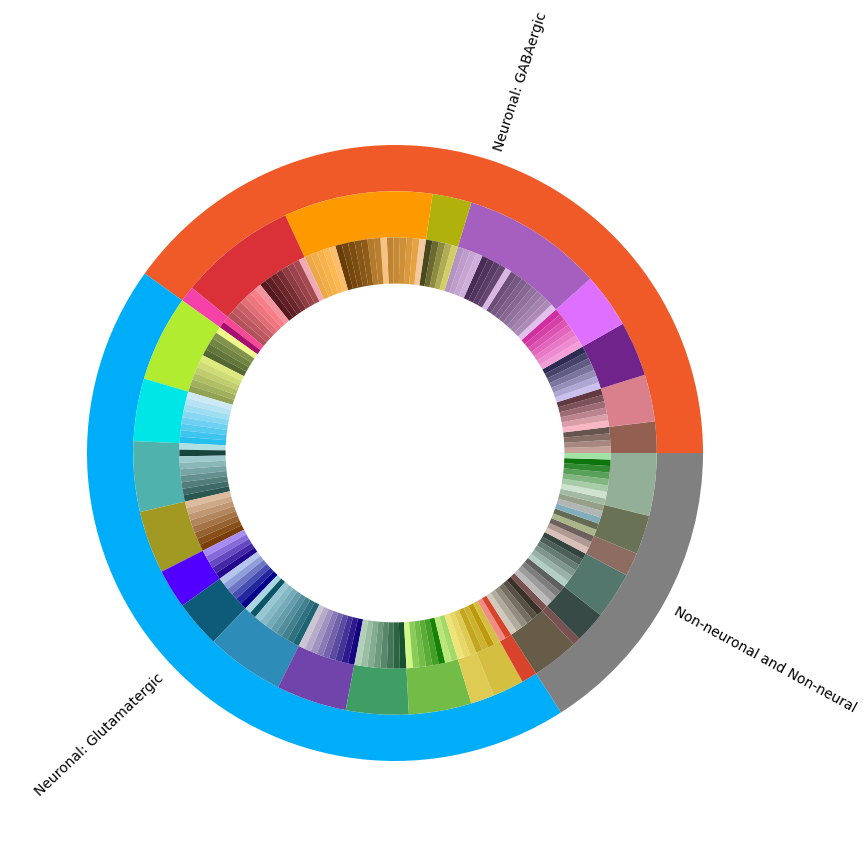
Term sets: Class, Subclass, and Supertype define the SEA-AD Multiregion taxonomy. We can visualize the taxonomy as a sunburst diagram that shows the single inheritance hierarchy through a series of rings, that are sliced for each annotation term. Each ring corresponds to a level in the hierarchy. We have ordered the rings so that the class level. Rings are divided based on their hierarchical relationship to the parent slice.
levels = ['Class', 'Subclass', 'Supertype']
df = {}
# Copy the term order of the parent into each of the level below it.
if term_with_counts.index.name != 'cluster_annotation_term_label':
term_with_counts = term_with_counts.set_index('cluster_annotation_term_label')
term_with_counts['parent_order'] = ""
for idx, row in term_with_counts.iterrows():
if pd.isna(row['parent_term_label']):
continue
term_with_counts.loc[idx, 'parent_order'] = term_with_counts.loc[row['parent_term_label']]['term_order']
term_with_counts = term_with_counts.reset_index()
for lvl in levels:
pred = term_with_counts['cluster_annotation_term_set_name'] == lvl
df[lvl] = term_with_counts[pred]
df[lvl] = df[lvl].sort_values(['parent_order', 'term_order'])
fig, ax = plt.subplots()
fig.set_size_inches(10, 10)
size = 0.15
for i, lvl in enumerate(levels):
if lvl == 'Class':
ax.pie(df[lvl]['number_of_supertypes'],
colors=df[lvl]['color_hex_triplet'],
labels = df[lvl]['name'],
rotatelabels=True,
labeldistance=1.025,
radius=1,
wedgeprops=dict(width=size, edgecolor=None),
startangle=0)
else :
ax.pie(df[lvl]['number_of_supertypes'],
colors=df[lvl]['color_hex_triplet'],
radius=1-i*size,
wedgeprops=dict(width=size, edgecolor=None),
startangle=0)
term_with_counts = term_with_counts.set_index('cluster_annotation_term_label')
plt.show()
Alzheimer’s Disease metrics#
Below we show some example plots for a specific AD metric (in this case Overall AD neuropathological Change).
The function below returns a heatmap of the fraction of cells in a given pair of features (col_1, col_2) normalized by the total in number cells in the second feature given (col_2).
import matplotlib as mpl
def plot_heatmap(
df: pd.DataFrame,
col_1: str,
col_2: str,
fig_width: float = 8,
fig_height: float = 4,
vmax: float = None,
cmap: plt.cm = plt.cm.magma,
log=False
):
"""Plot a heatmap of the proportion of cells in each group defined by col_1 and col_2.
The values in the heatmap represent the proportion of cells in each group normalized by the
total number of cells in each column group (col_2).
Parameters
----------
df : pd.DataFrame
DataFrame containing cell metadata and gene expression values.
col_1 : str
Column name in df to group by for the rows of the heatmap.
col_2 : str
Column name in df to group by for the columns of the heatmap.
fig_width : float, optional
Width of the figure in inches.
fig_height : float, optional
Height of the figure in inches.
vmax : float, optional
Maximum value for the color scale. If None, it is set to the maximum value in the data.
cmap : matplotlib colormap, optional
Colormap to use for the heatmap.
log : bool, optional
If True, the values will be log-transformed before plotting.
Returns
-------
fig : matplotlib.figure.Figure
The figure object containing the heatmap.
ax : array of matplotlib.axes.Axes
The axes objects for each species.
"""
fig, ax = plt.subplots(1, 1)
fig.set_size_inches(fig_width, fig_height)
grouped = df.groupby([col_1, col_2])['cell_barcode'].count().unstack()
grouped = grouped.div(grouped.sum(axis=0), axis=1) # Normalize to proportions
vmin = grouped.min().min()
vmax = grouped.max().max()
if log:
vmin = np.log10(vmin)
vmax = np.log10(vmax)
norm = mpl.colors.Normalize(vmin=vmin, vmax=vmax)
sm = plt.cm.ScalarMappable(cmap=cmap, norm=norm)
cmap = sm.get_cmap()
col_1_order = df.groupby(col_1)[[f'{col_1}_order']].first()
grouped = grouped.loc[
col_1_order.sort_values(f'{col_1}_order').index
]
col_2_order = df.groupby(col_2)[[f'{col_2}_order']].first()
grouped = grouped[
col_2_order.sort_values(f'{col_2}_order').index
]
arr = grouped.to_numpy().astype('float')
if log:
arr = np.log10(arr)
ax.imshow(arr, cmap=cmap, aspect='auto', vmin=vmin, vmax=vmax)
xlabs = grouped.columns.values
ylabs = grouped.index.values
ax.set_yticks(range(len(ylabs)))
ax.set_yticklabels(ylabs)
ax.set_xticks(range(len(xlabs)))
ax.set_xticklabels(xlabs)
cbar = fig.colorbar(sm, ax=ax, orientation='vertical', fraction=0.01, pad=0.01)
cbar.set_label('Proportion of Cells')
plt.subplots_adjust(wspace=0.00, hspace=0.00)
return fig, ax
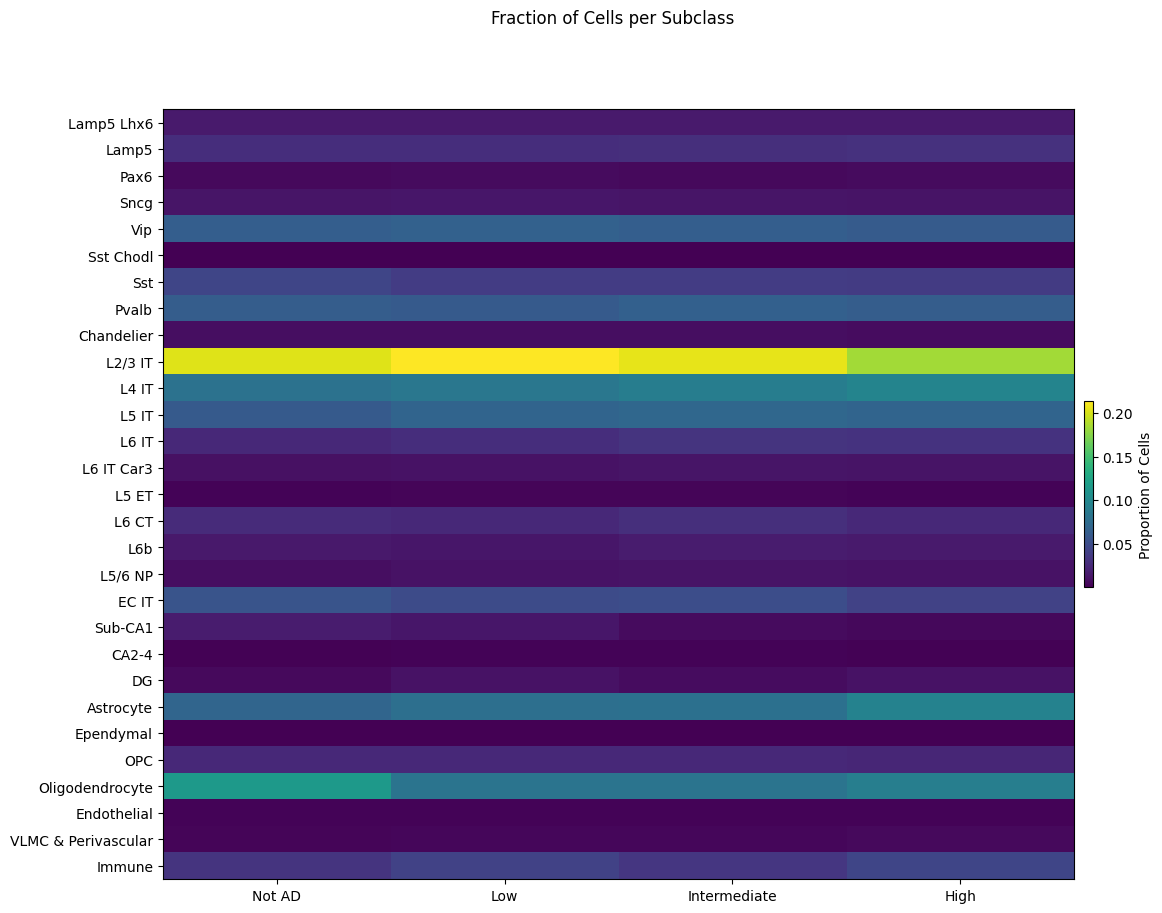
Below we plot the fraction of cells in a given Subclass across AD Neuropathological Change. Here we can see how the proportion of cell types changes as the AD Metric advances.
fig, ax = plot_heatmap(
df=cell_extended,
col_1="Subclass",
col_2="Overall AD neuropathological Change",
fig_width=12,
fig_height=10,
cmap=plt.cm.viridis
)
fig.suptitle('Fraction of Cells per Subclass')
plt.show()
In the next tutorial, we show how to access and use SEA-AD Mutliregion gene expression data. You can also investigate the SEA-AD Caudate data and taxonomy here.