Human-Mammalian Brain - Basal Ganglia 10X snRANSeq analysis: gene expression#
In this notebook we’ll explore some gene expressions and combine them with the cell metadata we showed in the previous clustering analysis tutorial.
You need to be connected to the internet to run this notebook or connected to a cache that has the Human-Mammalian Brain Atlas - Basal Ganglia (HMBA-BG) data downloaded already.
The notebook presented here shows quick visualizations from precomputed metadata in the atlas. For examples on accessing the expression matrices, specifically selecting genes from expression matrices, see the general Accessing expression data tutorial.
import pandas as pd
import numpy as np
from pathlib import Path
import matplotlib.pyplot as plt
from typing import List, Optional
from abc_atlas_access.abc_atlas_cache.abc_project_cache import AbcProjectCache
from abc_atlas_access.abc_atlas_cache.anndata_utils import get_gene_data
We will interact with the data using the AbcProjectCache. This cache object tracks which data has been downloaded and serves the path to the requested data on disk. For metadata, the cache can also directly serve up a Pandas DataFrame. See the getting_started notebook for more details on using the cache including installing it if it has not already been.
Change the download_base variable to where you would like to download the data to your system.
download_base = Path('../../data/abc_atlas')
abc_cache = AbcProjectCache.from_cache_dir(
download_base,
)
abc_cache.current_manifest
'releases/20260331/manifest.json'
Create the expanded cell metadata as was done previously in the cluster annotation tutorial tutorial.
# Load the cell metadata.
cell = abc_cache.get_metadata_dataframe(
directory='HMBA-10xMultiome-BG-Aligned',
file_name='cell_metadata',
dtype={'cell_label': str}
).set_index('cell_label')
donor = abc_cache.get_metadata_dataframe(
directory='HMBA-10xMultiome-BG-Aligned',
file_name='donor'
).set_index('donor_label')
library = abc_cache.get_metadata_dataframe(
directory='HMBA-10xMultiome-BG-Aligned',
file_name='library'
).set_index('library_label')
value_sets = abc_cache.get_metadata_dataframe(
directory='HMBA-10xMultiome-BG-Aligned',
file_name='value_sets'
).set_index('label')
cell_extended = cell.join(donor, on='donor_label')
cell_extended = cell_extended.join(
library, on='library_label',
rsuffix='_library_table'
)
def extract_value_set(cell_metadata_df: pd.DataFrame, input_value_set: pd.DataFrame, input_value_set_label: str):
"""Add color and order columns to the cell metadata dataframe based on the input
value set.
Columns are added as {input_value_set_label}_color and {input_value_set_label}_order.
Parameters
----------
cell_metadata_df : pd.DataFrame
DataFrame containing cell metadata.
input_value_set : pd.DataFrame
DataFrame containing the value set information.
input_value_set_label : str
The the column name to extract color and order information for. will be added to the cell metadata.
"""
cell_metadata_df[f'{input_value_set_label}_color'] = input_value_set[
input_value_set['field'] == input_value_set_label
].loc[cell_metadata_df[input_value_set_label]]['color_hex_triplet'].values
cell_metadata_df[f'{input_value_set_label}_order'] = input_value_set[
input_value_set['field'] == input_value_set_label
].loc[cell_metadata_df[input_value_set_label]]['order'].values
extract_value_set(cell_extended, value_sets, 'region_of_interest_label')
extract_value_set(cell_extended, value_sets, 'species_genus')
extract_value_set(cell_extended, value_sets, 'species_scientific_name')
extract_value_set(cell_extended, value_sets, 'donor_sex')
# Load the cluster memembership metadata and combine the data with the cell data.
cell_2d_embedding_coordinates = abc_cache.get_metadata_dataframe(
directory='HMBA-BG-taxonomy-CCN20250428',
file_name='cell_2d_embedding_coordinates'
).set_index('cell_label')
cell_extended = cell_extended.join(cell_2d_embedding_coordinates)
cell_extended = cell_extended.sample(frac=1)
cell_to_cluster_membership = abc_cache.get_metadata_dataframe(
directory='HMBA-BG-taxonomy-CCN20250428',
file_name='cell_to_cluster_membership',
).set_index('cell_label')
cluster = abc_cache.get_metadata_dataframe(
directory='HMBA-BG-taxonomy-CCN20250428',
file_name='cluster',
dtype={'number_of_cells': 'Int64'}
).rename(columns={'label': 'cluster_annotation_term_label'}).set_index('cluster_annotation_term_label')
cluster_annotation_term_set = abc_cache.get_metadata_dataframe(
directory='HMBA-BG-taxonomy-CCN20250428',
file_name='cluster_annotation_term_set'
).rename(columns={'label': 'cluster_annotation_term_label'})
cluster_annotation_term = abc_cache.get_metadata_dataframe(
directory='HMBA-BG-taxonomy-CCN20250428',
file_name='cluster_annotation_term',
).rename(columns={'label': 'cluster_annotation_term_label'}).set_index('cluster_annotation_term_label')
cell_to_cluster_membership = cell_to_cluster_membership[cell_to_cluster_membership['cluster_label'].isin(cluster_annotation_term.index)]
cluster_to_cluster_annotation_membership = abc_cache.get_metadata_dataframe(
directory='HMBA-BG-taxonomy-CCN20250428',
file_name='cluster_to_cluster_annotation_membership',
).set_index('cluster_annotation_term_label')
membership_with_cluster_info = cluster_to_cluster_annotation_membership.join(
cluster.reset_index().set_index('cluster_alias')[['number_of_cells']],
on='cluster_alias'
)
membership_with_cluster_info = cluster_to_cluster_annotation_membership.join(cluster_annotation_term, rsuffix='_anno_term').reset_index()
membership_groupby = membership_with_cluster_info.groupby(
['cluster_alias', 'cluster_annotation_term_set_name']
)
cluster_annotation_term_set = abc_cache.get_metadata_dataframe(
directory='HMBA-BG-taxonomy-CCN20250428',
file_name='cluster_annotation_term_set'
).rename(columns={'label': 'cluster_annotation_term_label'})
cluster_details = membership_groupby['cluster_annotation_term_name'].first().unstack()
cluster_order = membership_groupby['term_order'].first().unstack()
cluster_order.sort_values(['Neighborhood', 'Class', 'Subclass', 'Group', 'Cluster'], inplace=True)
cluster_order.rename(
columns={'Neighborhood': 'Neighborhood_order',
'Class': 'Class_order',
'Subclass': 'Subclass_order',
'Group': 'Group_order',
'Cluster': 'Cluster_order'},
inplace=True
)
cluster_colors = membership_groupby['color_hex_triplet'].first().unstack()
cluster_colors = cluster_colors[cluster_annotation_term_set['name']]
cluster_colors.sort_values(
['Neighborhood', 'Class', 'Subclass', 'Group', 'Cluster'],
inplace=True
)
cell_extended = cell_extended.join(cell_to_cluster_membership, how='inner')
cell_extended = cell_extended[~pd.isna(cell_extended['cluster_alias'])]
cell_extended = cell_extended.join(cluster_details, on='cluster_alias')
cell_extended = cell_extended.join(cluster_colors, on='cluster_alias', rsuffix='_color')
cell_extended = cell_extended.join(cluster_order, on='cluster_alias')
cell_extended.head(5)
/Users/chris.morrison/src/abc_atlas_access/src/abc_atlas_access/abc_atlas_cache/abc_project_cache.py:643: DtypeWarning: Columns (5) have mixed types. Specify dtype option on import or set low_memory=False.
return pd.read_csv(path, **kwargs)
| cell_barcode | donor_label | barcoded_cell_sample_label | library_label | alignment_job_id | doublet_score | umi_count | feature_matrix_label | dataset_label | abc_sample_id | ... | Neighborhood_color | Class_color | Subclass_color | Group_color | Cluster_color | Class_order | Cluster_order | Group_order | Neighborhood_order | Subclass_order | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell_label | |||||||||||||||||||||
| AGTAACCTCCTTAATC-1697_C04 | AGTAACCTCCTTAATC | QM23.50.001 | 1697_C04 | L8XR_230505_01_A08 | 0cac73b6e5f54f68d219e6e1449ad26964c65fd6 | 0.00 | 5161.0 | HMBA-10xMultiome-BG-Aligned | HMBA-10xMultiome-BG-Aligned | d305088f-31f5-46b5-b788-1a226d3f4e54 | ... | #a8afa5 | #401e66 | #401e66 | #195f8d | #5afd90 | 1 | 21 | 1 | 1 | 1 |
| ATCGCCCGTAACAGGG-2229_A06 | ATCGCCCGTAACAGGG | H24.30.004 | 2229_A06 | L8XR_240516_21_D12 | 257acbad4daec630edb9e1275971e151093a9b97 | 0.05 | 7171.0 | HMBA-10xMultiome-BG-Aligned | HMBA-10xMultiome-BG-Aligned | 5b3172d0-bfef-45c4-9a14-061f47cf0bf2 | ... | #a8afa5 | #401e66 | #401e66 | #195f8d | #3534d4 | 1 | 17 | 1 | 1 | 1 |
| TTTCTTGCAGCAACCT-2229_A06 | TTTCTTGCAGCAACCT | H24.30.004 | 2229_A06 | L8XR_240516_21_D12 | 257acbad4daec630edb9e1275971e151093a9b97 | 0.10 | 2115.0 | HMBA-10xMultiome-BG-Aligned | HMBA-10xMultiome-BG-Aligned | 71394bfb-3f99-402f-a3c3-fba115aeb674 | ... | #a8afa5 | #401e66 | #401e66 | #195f8d | #e1706a | 1 | 6 | 1 | 1 | 1 |
| AGAGAAGCACTAAATC-2459_B05 | AGAGAAGCACTAAATC | H21.30.004 | 2459_B05 | L8XR_240926_21_G09 | 9df076b587ec0d9bfbd6e52976ebccfad78972a7 | 0.00 | 4590.0 | HMBA-10xMultiome-BG-Aligned | HMBA-10xMultiome-BG-Aligned | 16e67220-9fb6-49d9-a02a-425b7ae2a341 | ... | #a8afa5 | #594a26 | #594a26 | #66391F | #a9de1a | 3 | 142 | 9 | 1 | 7 |
| GATGACTTCCCGCATT-2289_D04 | GATGACTTCCCGCATT | H24.30.003 | 2289_D04 | L8XR_240627_21_A02 | 65dd629a0037be17954a4fe363a62a5de2075478 | 0.16 | 7003.0 | HMBA-10xMultiome-BG-Aligned | HMBA-10xMultiome-BG-Aligned | ab93f81c-7094-4354-a745-62dff72b55b4 | ... | #19613b | #d0b83c | #253c8c | #aec7e8 | #3d70fc | 10 | 1178 | 53 | 3 | 32 |
5 rows × 59 columns
Single cell transcriptomes#
The ~2 million, 10X single cell dataset of HMBA-BG is available in two separate packages. The first contains a single aligned h5ad file containing ~16k genes that have been aligned across all species. We use this single aligned dataset in this notebook. The other package is split across each species with a separate h5ad file for each species. These individually containing roughly ~36k genes for each species.
Below we show some interactions with data from the 10X expression matrices in the HMBA-BG dataset. For a deeper dive into how to access specific gene data from the expression matrices, take a look at general accessing expression data tutorial.
First, we list the available metadata in the HMBA-BG 10X dataset again.
abc_cache.list_metadata_files('HMBA-10xMultiome-BG-Aligned')
['cell_metadata',
'donor',
'example_gene_expression',
'gene',
'library',
'value_sets']
For completeness, we list the available expression matrix files.
abc_cache.list_expression_matrix_files('HMBA-10xMultiome-BG-Aligned')
['HMBA-10xMultiome-BG-Aligned/log2', 'HMBA-10xMultiome-BG-Aligned/raw']
We first load the gene data for the Aligned dataset covering all species in the BG dataset.
aligned_gene = abc_cache.get_metadata_dataframe(
directory='HMBA-10xMultiome-BG-Aligned',
file_name='gene'
).set_index('gene_identifier')
print("Number of aligned genes = ", len(aligned_gene))
aligned_gene.head(5)
gene.csv: 100%|██████████| 1.15M/1.15M [00:00<00:00, 5.17MMB/s]
Number of aligned genes = 16630
| gene_symbol | description | molecular_type | |
|---|---|---|---|
| gene_identifier | |||
| NCBIGene:9380 | GRHPR | glyoxylate and hydroxypyruvate reductase | protein-coding |
| NCBIGene:6603 | SMARCD2 | SWI/SNF related, matrix associated, actin depe... | protein-coding |
| NCBIGene:148103 | ZNF599 | zinc finger protein 599 | protein-coding |
| NCBIGene:92691 | TMEM169 | transmembrane protein 169 | protein-coding |
| NCBIGene:3235 | HOXD9 | homeobox D9 | protein-coding |
Below we list the genes we will use in this notebook and the example method used to load the expression for these specific genes from the h5ad file. Note that we provide a set of example gene expressions as a csv file for brevity in this tutorial. To process and extract the gene expressions for yourself, uncomment the code block below. More details on how to extract specific genes from the data see our accessing gene expression data tutorial
gene_names = ['SLC17A6', 'SLC32A1', 'PTPRC', 'PLP1', 'AQP4', 'DRD1', 'DRD2']
"""
aligned_gene_data = get_gene_data(
abc_atlas_cache=abc_cache,
all_cells=cell_extended,
all_genes=aligned_gene,
selected_genes=gene_names
)
"""
'\naligned_gene_data = get_gene_data(\n abc_atlas_cache=abc_cache,\n all_cells=cell_extended,\n all_genes=aligned_gene,\n selected_genes=gene_names\n)\n'
Instead of processing the gene expressions, we load a pre-processed file.
aligned_gene_data = abc_cache.get_metadata_dataframe(
'HMBA-10xMultiome-BG-Aligned',
'example_gene_expression'
).set_index('cell_label')
example_gene_expression.csv: 100%|██████████| 115M/115M [00:08<00:00, 13.5MMB/s]
Next, we’ll concatenate the gene data together and merge them into our cell metadata.
cell_extended_with_genes = cell_extended.join(aligned_gene_data)
Example use cases#
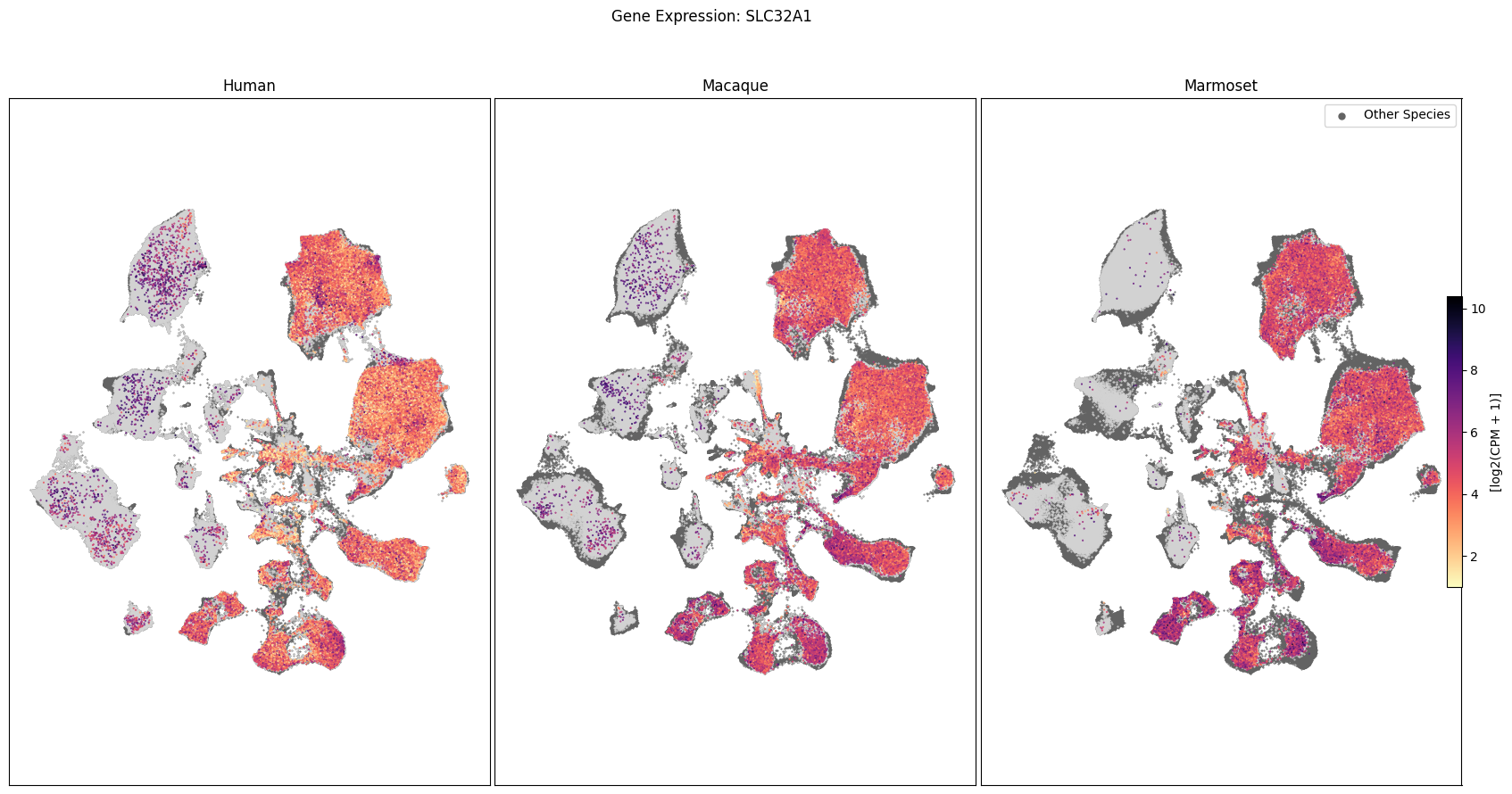
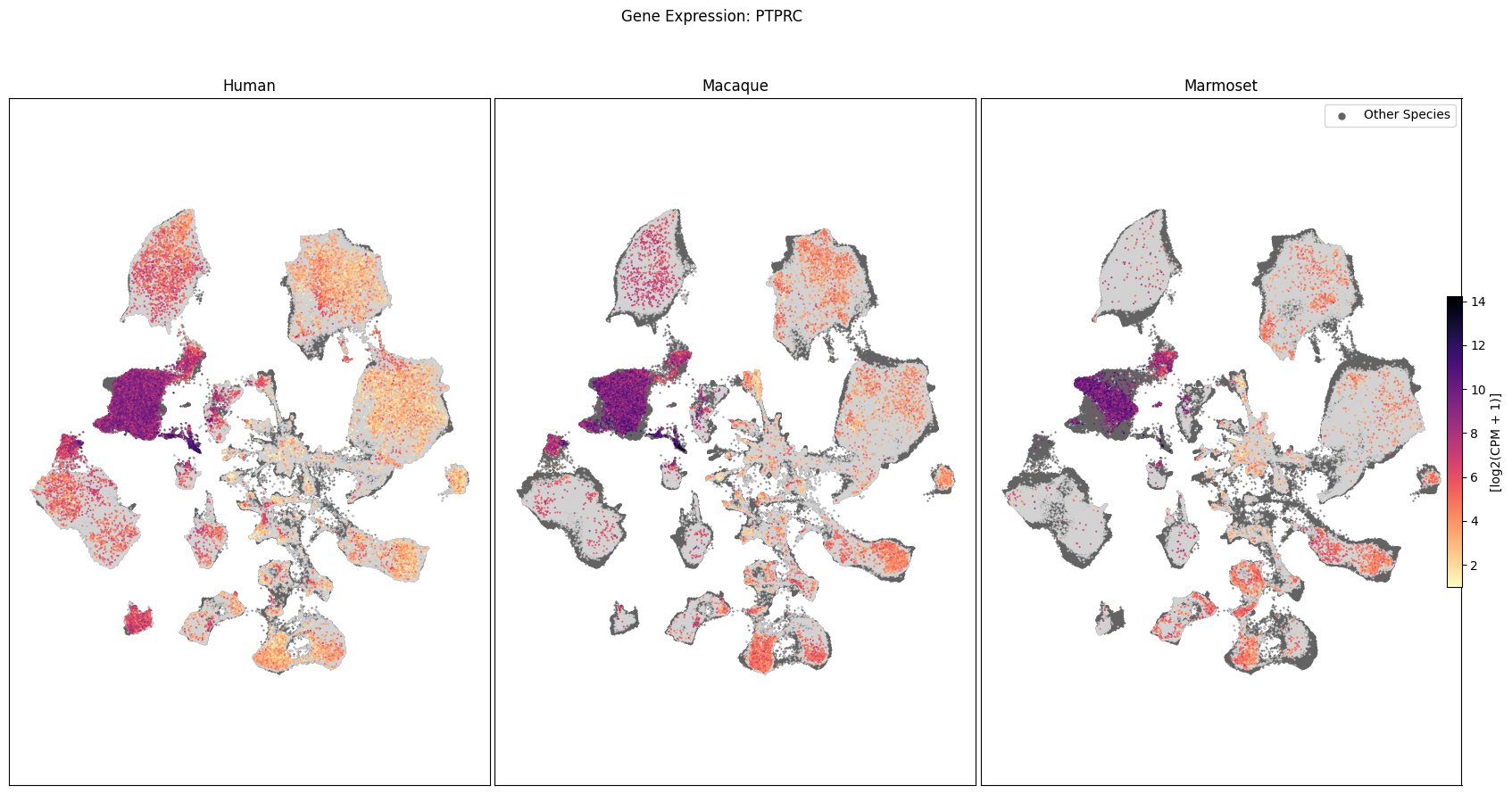
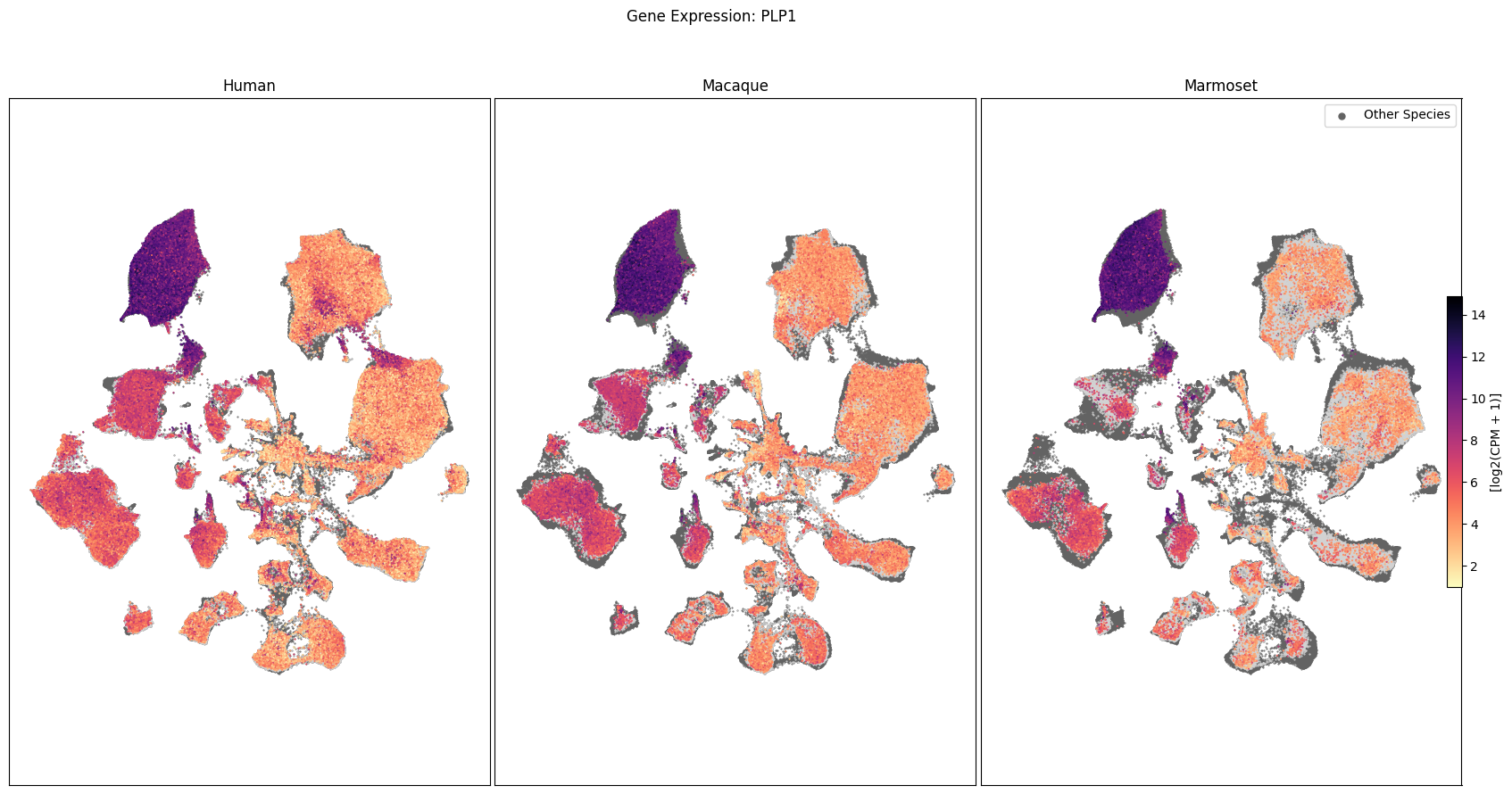
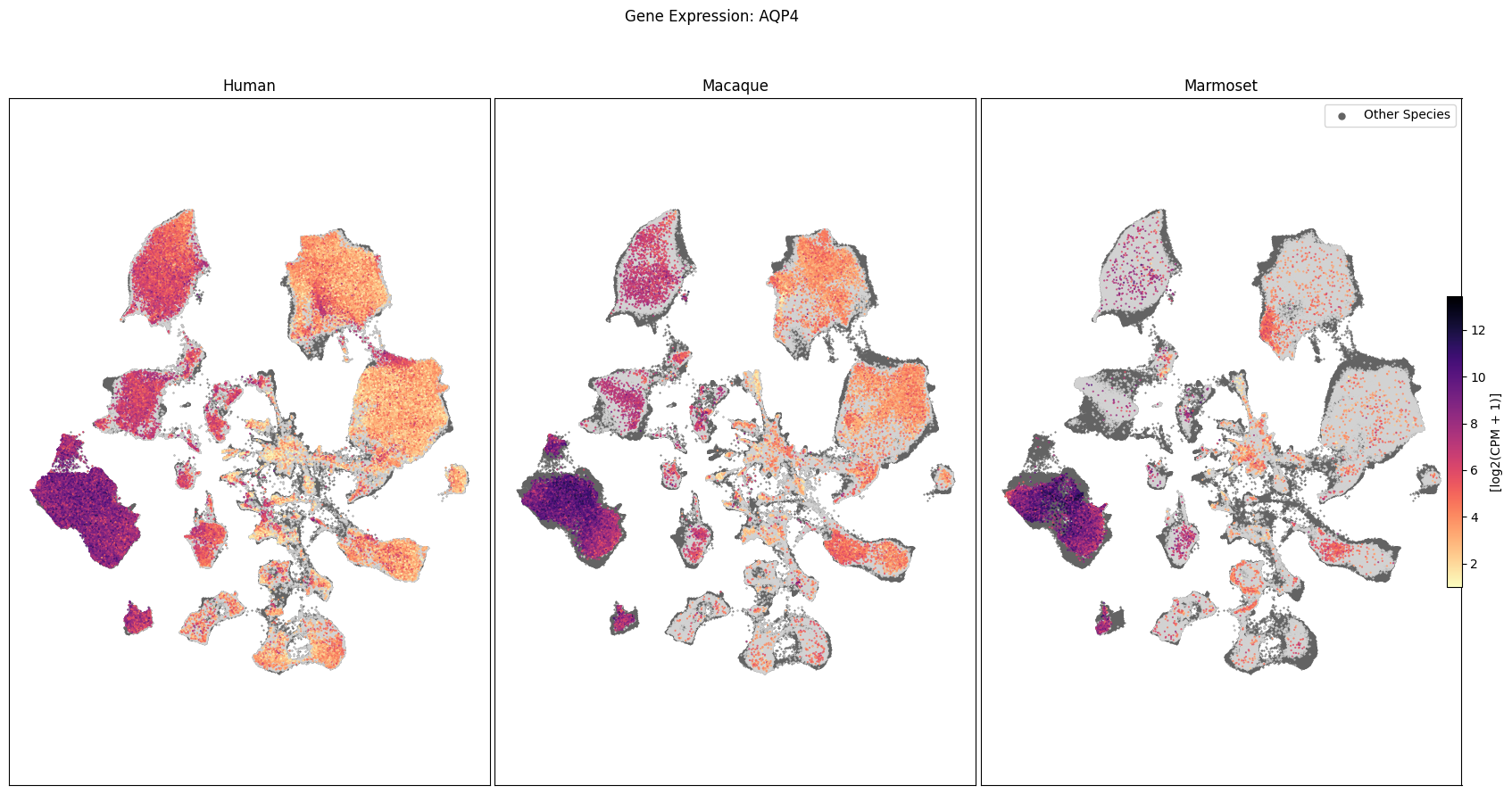
In this section, we show a use case with the example genes SLC17A6, SLC32A1, PTPRC, PLP1, AQP4, DRD1, and DRD2. These genes were selected because they are marker genes for glutamatergic (SLC17A6) and GABAergic (SLC32A1) neurons, immune cells (PTPRC), oligodendrocytes (PLP1), astrocytes (AQP4), and D1/D2 medium spiny neurons (DRD1, DRD2). “Marker genes” have much higher expression in the specified cell type or anatomic structure when compared to all other cells, and in many cases are functionally relevant for those cell types.
The helper function below creates a heatmap showing the relation between various parameters in the combined cell metadata and the genes we loaded.
import matplotlib as mpl
def plot_species_heatmap(
df: pd.DataFrame,
gnames: List[str],
value: str,
species_list: List[str] = None,
sort: bool = False,
fig_width: float = 8,
fig_height: float = 4,
vmax: float = None,
cmap: plt.cm = plt.cm.magma_r
):
"""Plot a heatmap of gene expression values for a list of genes across species.
Parameters
----------
df : pd.DataFrame
DataFrame containing cell metadata and gene expression values.
gnames : list
List of gene names to plot.
value : str
Column name in df to group by (e.g., 'species_genus').
species_list : list, optional
List of species to include in the plot. If None, all unique species in df are used.
sort : bool, optional
Whether to sort the gene expression values within each species.
fig_width : float, optional
Width of the figure in inches.
fig_height : float, optional
Height of the figure in inches.
vmax : float, optional
Maximum value for the color scale. If None, it is set to the maximum value in the data.
cmap : matplotlib colormap, optional
Colormap to use for the heatmap.
Returns
-------
fig : matplotlib.figure.Figure
The figure object containing the heatmap.
ax : array of matplotlib.axes.Axes
The axes objects for each species.
"""
fig, ax = plt.subplots(1, len(species_list))
fig.set_size_inches(fig_width, fig_height)
all_unique_values = df[value].unique()
try:
order = df[f'{value}_order'].unique()
all_unique_values = all_unique_values[np.argsort(order)]
except KeyError:
order = None
all_grouped = df.groupby(value)[gnames].mean()
vmin = all_grouped.min().min()
vmax = all_grouped.max().max()
norm = mpl.colors.Normalize(vmin=vmin, vmax=vmax)
sm = plt.cm.ScalarMappable(cmap=cmap, norm=norm)
cmap = sm.get_cmap()
for idx, species in enumerate(species_list):
filtered = df[df['species_genus'] == species]
grouped = filtered.groupby(value)[gnames].mean()
if sort:
grouped = grouped.sort_values(by=gnames[0], ascending=False)
missing_values = []
indices = []
for unique_value in all_unique_values:
if unique_value not in grouped.index:
indices.append(unique_value)
missing_values.append({key: np.nan for key in gnames})
if missing_values:
grouped = pd.concat([grouped, pd.DataFrame(data=missing_values, index=indices)])
grouped = grouped.loc[all_unique_values]
arr = grouped.to_numpy().astype('float')
ax[idx].imshow(arr, cmap=cmap, aspect='auto', vmin=vmin, vmax=vmax)
xlabs = grouped.columns.values
ylabs = grouped.index.values
if idx == 0:
ax[idx].set_yticks(range(len(ylabs)))
ax[idx].set_yticklabels(ylabs)
else:
ax[idx].set_yticks([])
ax[idx].set_yticklabels([])
ax[idx].set_xticks(range(len(xlabs)))
ax[idx].set_xticklabels(xlabs)
ax[idx].set_title(species)
cbar = fig.colorbar(sm, ax=ax, orientation='vertical', fraction=0.01, pad=0.01)
cbar.set_label('Mean Expression [log2(CPM + 1)]')
plt.subplots_adjust(wspace=0.00, hspace=0.00)
return fig, ax
Expression of selected genes in the Basal Ganglia#
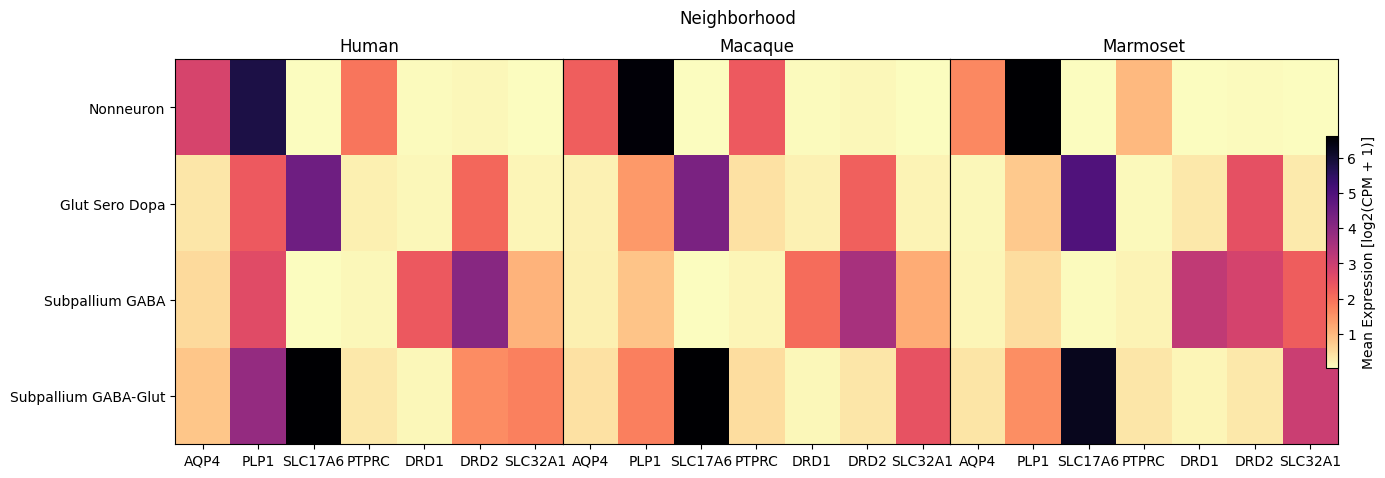
Below we use our heatmap function to plot the gene expression as a function of a feature in our data. For the first two plots, we show the expression for all species against the Neighborhood and Group levels of the taxonomy. Note that a band of white denotes a Group (or other feature) that is not present in for the given species.
fig, ax = plot_species_heatmap(
df=cell_extended_with_genes,
gnames=aligned_gene_data.columns,
value='Neighborhood',
species_list=['Human', 'Macaque', 'Marmoset'],
fig_width=15,
fig_height=5
)
fig.suptitle('Neighborhood')
plt.show()
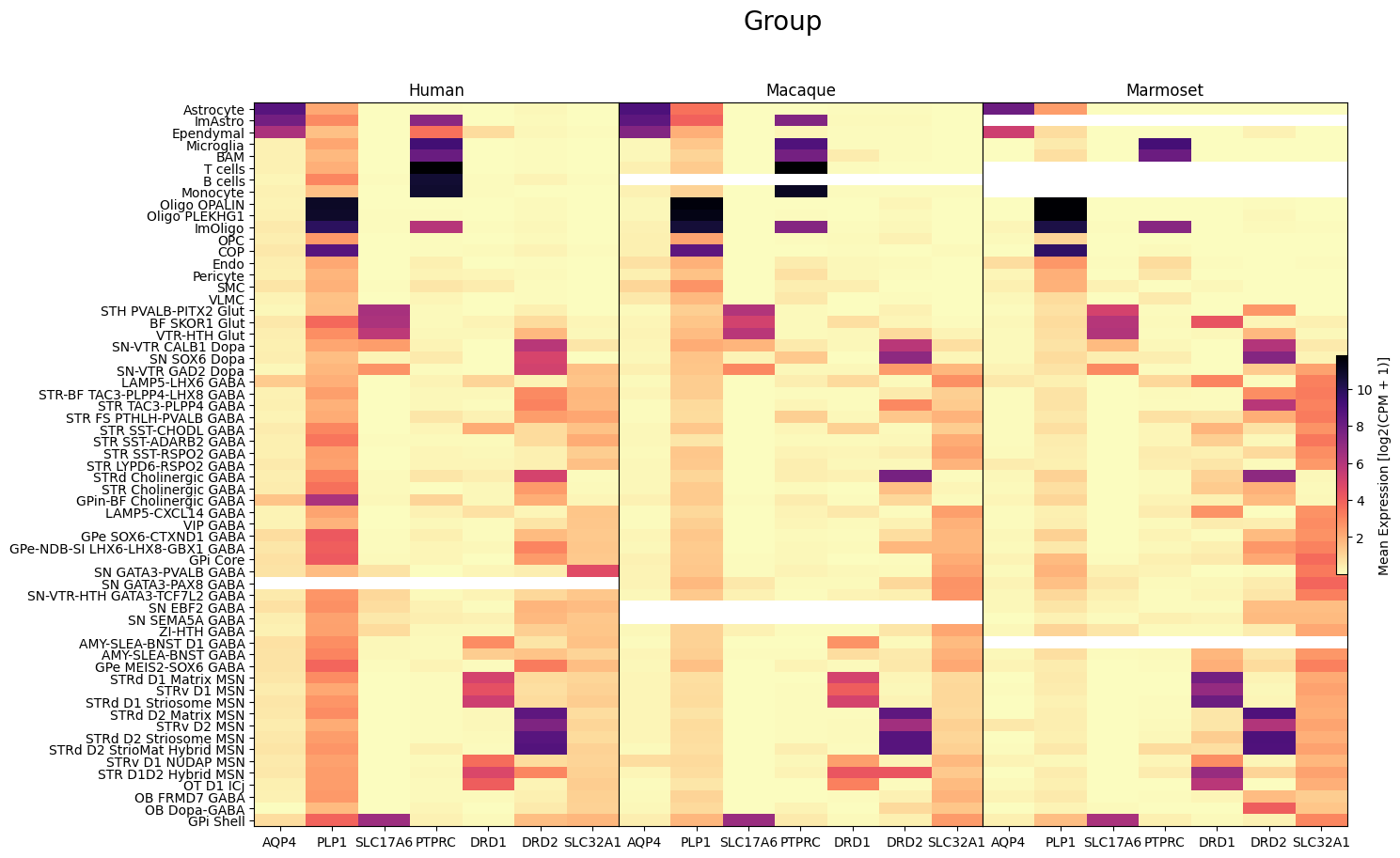
fig, ax = plot_species_heatmap(
df=cell_extended_with_genes,
gnames=aligned_gene_data.columns,
value='Group',
species_list=['Human', 'Macaque', 'Marmoset'],
fig_width=15,
fig_height=10
)
fig.suptitle('Group', size=20)
plt.show()
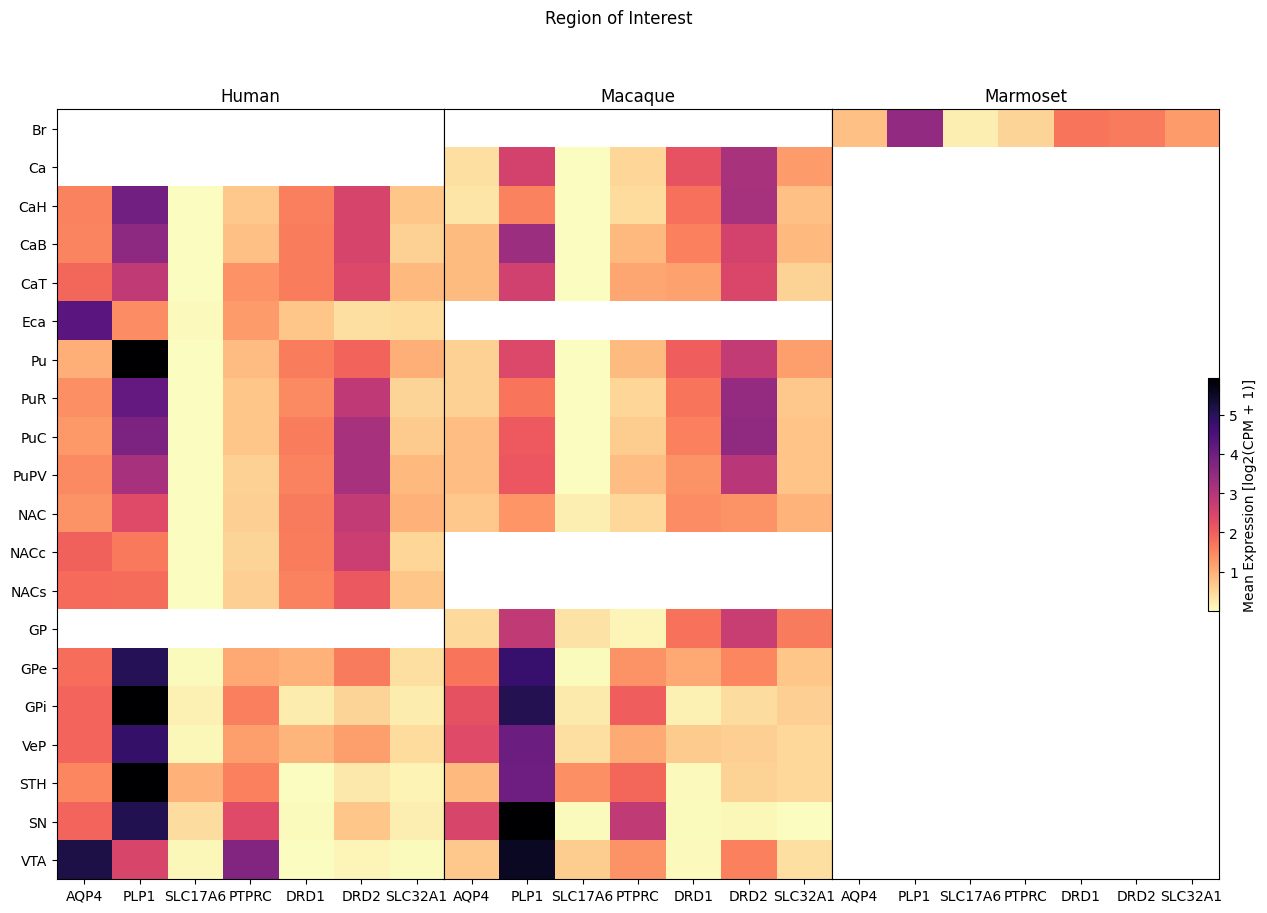
Now we perform the same heatmap comparison against the region of interest in the brain the cell was extracted from. Note that currently the Marmoset has no fine grained identification of brain region.
fig, ax = plot_species_heatmap(
df=cell_extended_with_genes,
gnames=aligned_gene_data.columns,
species_list=['Human', 'Macaque', 'Marmoset'],
value='region_of_interest_label',
fig_width=15,
fig_height=10
)
fig.suptitle('Region of Interest')
plt.show()
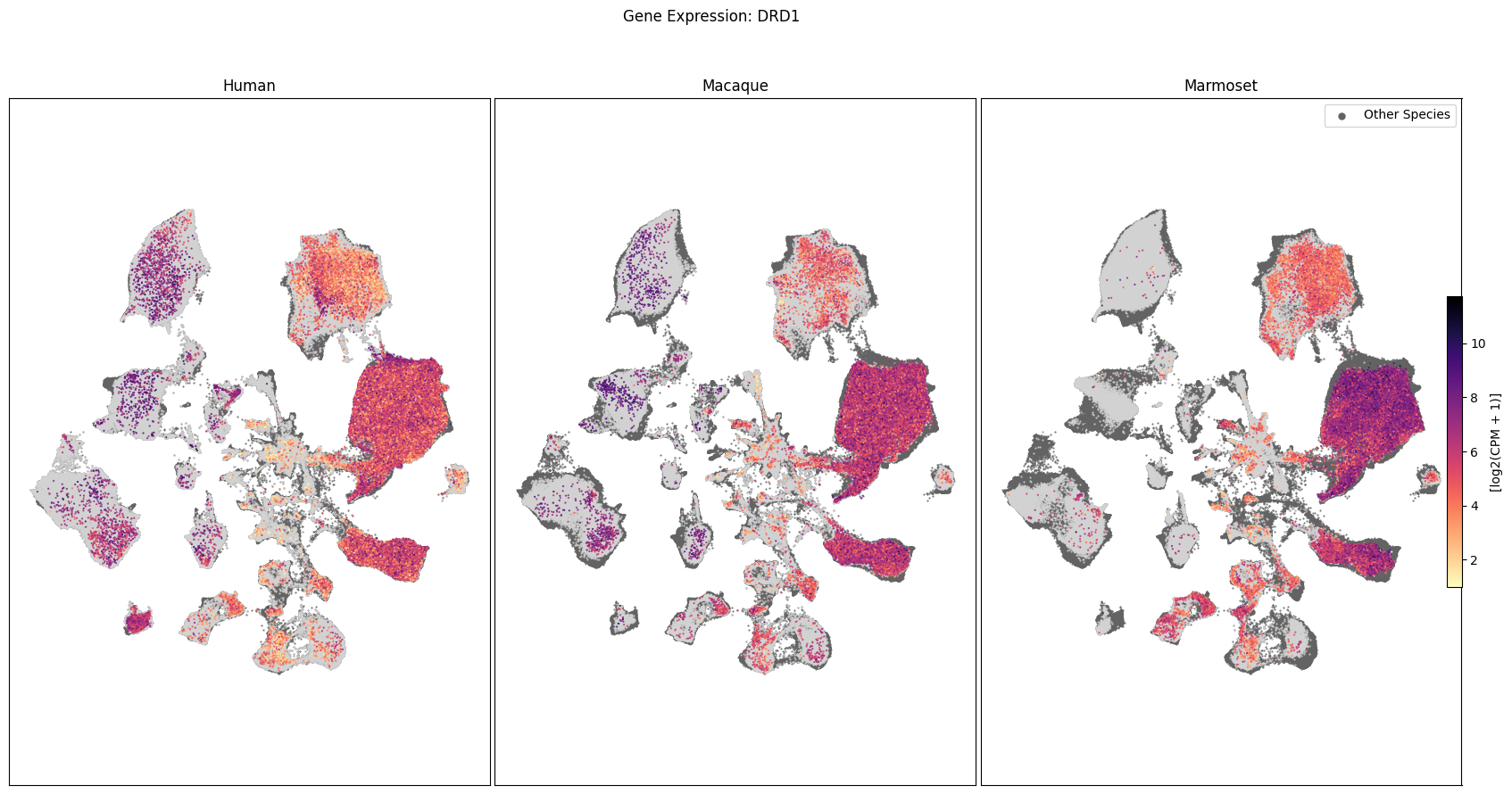
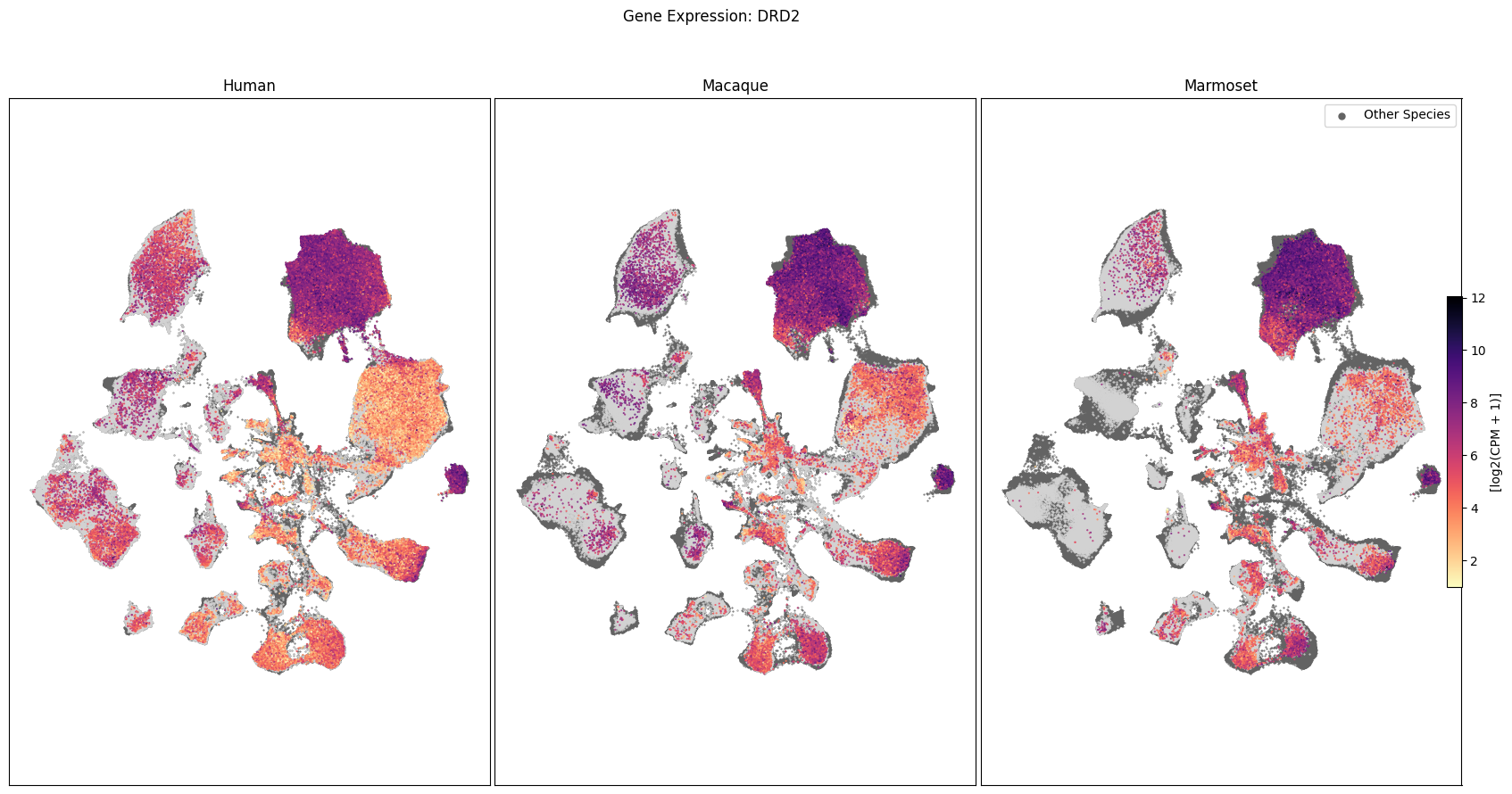
Expression in the UMAP#
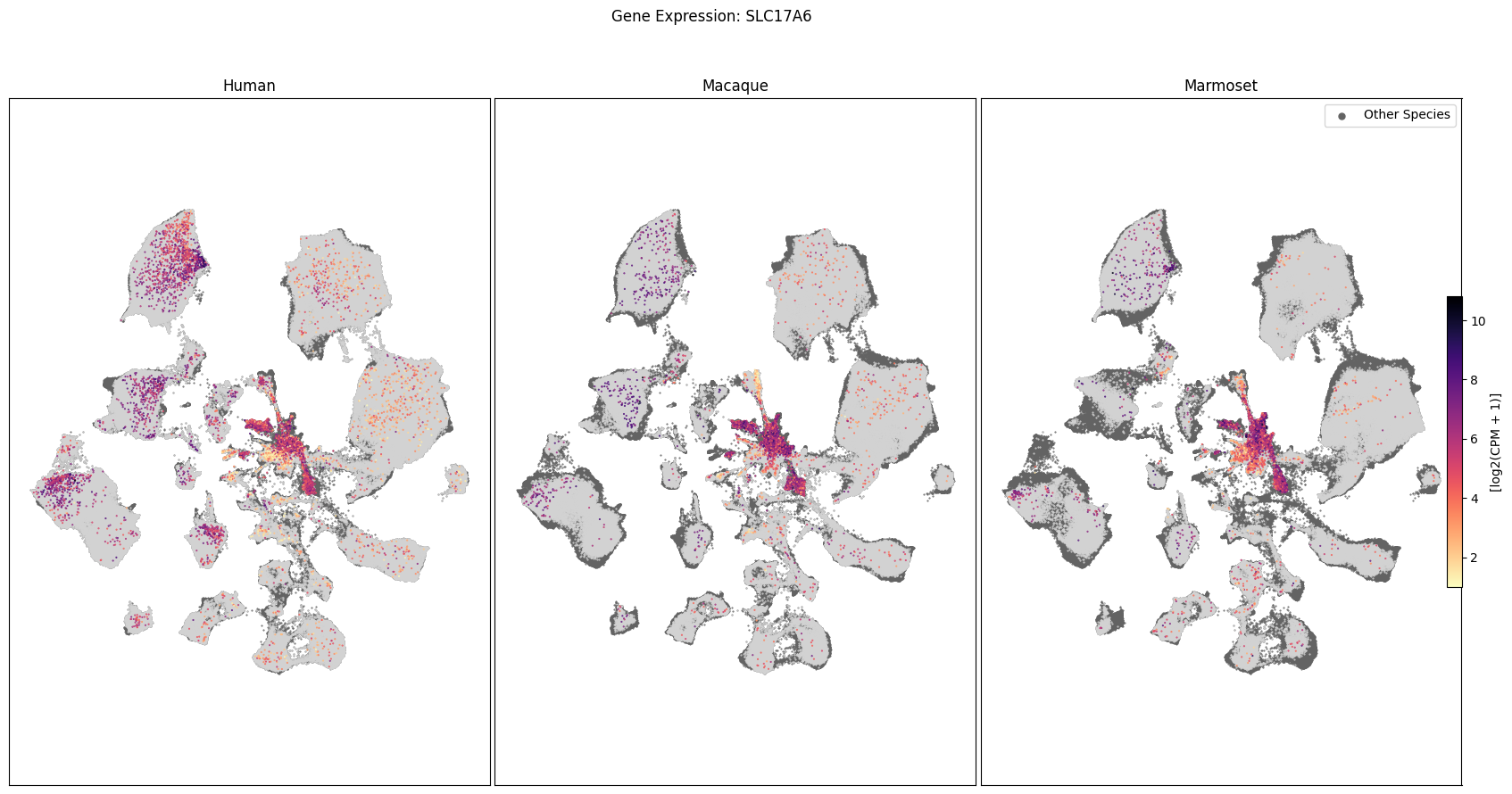
We can also visualize the relationship between these genes and their location in the UMAP. We’ll plot the species side by side with the dark grey cells in each plot representing those from the other species that do not overlap with the plotted one. We select a minium expression value of which effectively masks out zero expression cells from the plots, showing a bit more contrast. The cells below are selected value are plotted as a light grey. Feel free to experiment with different vmin and vmax values in investigate the gene expressions more in depth.
def plot_umap(
df: pd.DataFrame,
feature: str,
species_list: List[str],
cmap=None,
vmin: Optional[float] = None,
vmax: Optional[float] = None,
fig_width=21,
fig_height=10
):
"""Plot UMAP scatter plots for a given feature across multiple species.
Parameters
----------
df : pd.DataFrame
DataFrame containing cell metadata including 'x', 'y', and 'species_genus' columns.
feature : str
Column name in df to color the points by (e.g., gene expression).
species_list : list
List of species to include in the plot.
cmap : matplotlib colormap, optional
Colormap to use for coloring the points. If None, uses the feature values as colors directly.
vmin: float, optional
Minimum value for color mapping. Defaults to min of values in the feature column. Cells with
expression below this value will be plotted as light grey.
vmax: float, optional
Maximum value for color mapping. Defaults to max of values in the feature column. Cells with
expression above this value will be plotted as light grey.
fig_width : float, optional
Width of the figure in inches.
fig_height : float, optional
Height of the figure in inches.
Returns
-------
fig : matplotlib.figure.Figure
The figure object containing the UMAP plots.
ax : array of matplotlib.axes.Axes
The axes objects for each species.
"""
fig, ax = plt.subplots(1, len(species_list))
fig.set_size_inches(fig_width, fig_height)
all_xx = df['x']
all_yy = df['y']
vv = df[feature]
if vmin is None:
vmin = vv.min()
if vmax is None:
vmax = vv.max()
norm = mpl.colors.Normalize(vmin=vmin, vmax=vmax)
sm = plt.cm.ScalarMappable(cmap=cmap, norm=norm)
cmap = sm.get_cmap()
for idx, species in enumerate(species_list):
species_mask = df['species_genus'] == species
filtered = df[species_mask]
xx = filtered['x']
yy = filtered['y']
vv = filtered[feature]
ax[idx].scatter(all_xx, all_yy, s=1.0, color='#636363', marker='.', label='Other Species')
if cmap is not None:
mask = np.logical_and(vv >= vmin, vv <= vmax)
if mask.any():
ax[idx].scatter(xx[~mask], yy[~mask], s=1.0, c='#D3D3D3', marker='.')
ax[idx].scatter(xx[mask], yy[mask], s=1.0, c=vv[mask], marker='.', cmap=cmap)
else:
ax[idx].scatter(xx, yy, s=1.0, color=vv, marker=".")
else :
ax[idx].scatter(xx, yy, s=1.0, color=vv, marker=".")
ax[idx].axis('equal')
ax[idx].set_xticks([])
ax[idx].set_yticks([])
ax[idx].set_title(f"{species}")
cbar = fig.colorbar(sm, ax=ax, orientation='vertical', fraction=0.01, pad=0.01)
cbar.set_label('[log2(CPM + 1)]')
plt.legend(loc=0, markerscale=10)
plt.subplots_adjust(wspace=0.01, hspace=0.01)
return fig, ax
for gene_name in gene_names:
fig, ax = plot_umap(
cell_extended_with_genes,
feature=gene_name,
species_list=['Human', 'Macaque', 'Marmoset'],
cmap=plt.cm.magma_r,
vmin=1,
vmax=None
)
fig.suptitle(f'Gene Expression: {gene_name}')
plt.show()
To learn more about the taxonomy and metadata used in this notebook see the HMBA-BG Clustering Analysis and Taxonomy notebook.