Whole Human Brain 10x RNA-seq clustering analysis and annotation#
The Whole Human Brain Atlas data 10X RNA-seq dataset contains ~3 million nuclei (more than 2 million sequenced neurons and almost 1 million non-neurons) from 100 locations across the forebrain, midbrain, and hindbrain. The integrated transcriptomic taxonomy contains 31 superclusters, 461 clusters, and 3313 subclusters. Full details of the dataset can be found in Siletti et al. 2023
The purpose of this notebook is to provide an overview of how cluster and cluster annotation information is represented through example use cases. Note that cluster in this notebook is an overloaded term as one of the levels is referred to as cluster while it can also refer to the general clustering of similar cells.
You need to be connected to the internet to run this notebook and have run through the getting started notebook.
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
from pathlib import Path
from abc_atlas_access.abc_atlas_cache.abc_project_cache import AbcProjectCache
We will interact with the data using the AbcProjectCache. This cache object tracks which data has been downloaded and serves the path to the requsted data on disk. For metadata, the cache can also directly serve a up a Pandas Dataframe. See the getting_started notebook for more details on using the cache including installing it if it has not already been.
Change the download_base variable to where you have downloaded the data in your system.
download_base = Path('../../data/abc_atlas')
abc_cache = AbcProjectCache.from_cache_dir(download_base)
abc_cache.current_manifest
'releases/20250531/manifest.json'
Data Overview#
Below we list the available metadata in the WHB-taxonomy directory.
abc_cache.list_metadata_files('WHB-taxonomy')
['cluster',
'cluster_annotation_term',
'cluster_annotation_term_set',
'cluster_to_cluster_annotation_membership']
Subclusters#
Each of the final set of 3313 subclusters is associated with an alias and label. Each row of the dataframe represents a subcluster as defined in Siletti et al. 2023. Each subcluster has a label (string that is unique in the database), cluster alias (in this case a simple integer) and the number of cells that has been grouped into the cluster.
cluster = abc_cache.get_metadata_dataframe(directory='WHB-taxonomy', file_name='cluster')
print(len(cluster))
cluster
cluster.csv: 100%|██████████| 84.8k/84.8k [00:00<00:00, 88.6kMB/s]
3313
| cluster_alias | number_of_cells | label | |
|---|---|---|---|
| 0 | 0 | 34 | CS202210140_494 |
| 1 | 1 | 220 | CS202210140_495 |
| 2 | 2 | 187 | CS202210140_496 |
| 3 | 3 | 246 | CS202210140_497 |
| 4 | 4 | 188 | CS202210140_498 |
| ... | ... | ... | ... |
| 3308 | 3308 | 140 | CS202210140_3802 |
| 3309 | 3309 | 138 | CS202210140_3803 |
| 3310 | 3310 | 85 | CS202210140_3804 |
| 3311 | 3311 | 93 | CS202210140_3805 |
| 3312 | 3312 | 74 | CS202210140_3806 |
3313 rows × 3 columns
Cluster annotation term sets#
Each level of classification is represented as a cluster annotation term set. Each term set consists of a set of ordered terms. Each term set has a label (human readable string that is unique in the database), a name, description and order among the term sets.
term_set = abc_cache.get_metadata_dataframe(directory='WHB-taxonomy', file_name='cluster_annotation_term_set')
term_set
| label | name | description | order | |
|---|---|---|---|---|
| 0 | CCN202210140_SUBC | subcluster | The finest level of cell type definition in th... | 2 |
| 1 | CCN202210140_CLUS | cluster | An intermediate level of cell type definitions... | 1 |
| 2 | CCN202210140_SUPC | supercluster | The top level of cell type definition in the h... | 0 |
| 3 | CCN202210140_NEUR | neurotransmitter | Neurotransmitter terms are assigned to cluster... | 3 |
We can use the pandas iterrows function to print out the description for each term set.
# print out description for each term_set
for tsindex, tsrow in term_set.iterrows():
print("%s:\n" % tsrow['name'])
print("%s\n" % tsrow['description'])
subcluster:
The finest level of cell type definition in the human whole brain taxonomy. It is defined by applying Louvain clustering independently for each cluster. Cells within a subcluster share similar characteristics and belong to the same cluster. In some cases subclusters are distinct, and in other cases represent cell states or gradients within the same cluster.
cluster:
An intermediate level of cell type definitions in the human whole brain taxonomy. It is defined by applying Louvain clustering independently for each supercluster. All cells within a subcluster belong to the same cluster. Clusters group together similar subclusters and are highly distinct from one another.
supercluster:
The top level of cell type definition in the human whole brain taxonomy. It is defined using Louvain clustering (Cytograph) with most superclusters determined by broad brain region and neurotransmitter type. All cells within a cluster belong to the same supercluster. Supercluster provides a broader characterization of cell types.
neurotransmitter:
Neurotransmitter terms are assigned to clusters based on the expression (or lack of expression) of one or more neurotransmitter-associated transporter or enzyme genes
Cluster annotation terms#
A cluster annotation term represents a group within a single level of classification. Each term object has a label (human readable string that is unique in the database), a name, which cluster annotation term set it belongs to and a parent or superclass if the term forms a hierarchy, an order within a term set and a color for visualization purposes.
term = abc_cache.get_metadata_dataframe(directory='WHB-taxonomy', file_name='cluster_annotation_term', keep_default_na=False)
term.head(5)
| label | name | cluster_annotation_term_set_label | parent_term_label | parent_term_set_label | term_set_order | term_order | cluster_annotation_term_set_name | color_hex_triplet | number_of_cells | description | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | CS202210140_476 | Upper-layer intratelencephalic | CCN202210140_SUPC | 0 | 0 | supercluster | #FEA7BA | 455006 | Upper-layer intratelencephalic | ||
| 1 | CS202210140_477 | Deep-layer intratelencephalic | CCN202210140_SUPC | 0 | 1 | supercluster | #426600 | 228467 | Deep-layer intratelencephalic | ||
| 2 | CS202210140_473 | Deep-layer near-projecting | CCN202210140_SUPC | 0 | 2 | supercluster | #EE1010 | 18856 | Deep-layer near-projecting | ||
| 3 | CS202210140_474 | Deep-layer corticothalamic and 6b | CCN202210140_SUPC | 0 | 3 | supercluster | #5EF0F1 | 78396 | Deep-layer corticothalamic and 6b | ||
| 4 | CS202210140_484 | MGE interneuron | CCN202210140_SUPC | 0 | 4 | supercluster | #DFFE66 | 222434 | MGE interneuron |
We can use the groupby and count functions in pandas to tally the number of terms in each term set (classification level).
term[['label','cluster_annotation_term_set_name']].groupby('cluster_annotation_term_set_name').count()
| label | |
|---|---|
| cluster_annotation_term_set_name | |
| cluster | 461 |
| neurotransmitter | 20 |
| subcluster | 3313 |
| supercluster | 31 |
Unlike the Whole Mouse brain data, the term names are unique across term sets.
Cluster to cluster annotation membership#
The association between a subcluster and cluster annotation term is represented as a cluster to cluster annotation membership within the context of one cluster annotation term set. It is expected that a cluster in only associated with one term within a specific term set.
membership = abc_cache.get_metadata_dataframe(directory='WHB-taxonomy', file_name='cluster_to_cluster_annotation_membership')
membership.head(5)
| cluster_annotation_term_label | cluster_annotation_term_set_label | cluster_alias | cluster_annotation_term_name | cluster_annotation_term_set_name | number_of_cells | color_hex_triplet | |
|---|---|---|---|---|---|---|---|
| 0 | CS202210140_494 | CCN202210140_SUBC | 0 | URL_297_0 | subcluster | 34 | #7E807A |
| 1 | CS202210140_495 | CCN202210140_SUBC | 1 | URL_308_1 | subcluster | 220 | #C54945 |
| 2 | CS202210140_496 | CCN202210140_SUBC | 2 | URL_308_2 | subcluster | 187 | #5232B7 |
| 3 | CS202210140_497 | CCN202210140_SUBC | 3 | URL_308_3 | subcluster | 246 | #31BEBA |
| 4 | CS202210140_498 | CCN202210140_SUBC | 4 | URL_308_4 | subcluster | 188 | #C8A9BC |
Example use cases#
Aggregating cluster and cells counts per term#
We can obtain cluster and cell counts per cluster annotation term using the pandas groupby function.
# Count the number of clusters associated with each cluster annotation term
term_cluster_count = membership.groupby(['cluster_annotation_term_label'])[['cluster_alias']].count()
term_cluster_count.columns = ['number_of_clusters']
term_cluster_count.head(5)
| number_of_clusters | |
|---|---|
| cluster_annotation_term_label | |
| CS202210140_1 | 1 |
| CS202210140_10 | 2 |
| CS202210140_100 | 9 |
| CS202210140_1000 | 1 |
| CS202210140_1001 | 1 |
# Join counts with the term dataframe
term_by_label = term.set_index('label')
term_with_counts = term_by_label.join(term_cluster_count)
term_with_counts[['name', 'cluster_annotation_term_set_name', 'number_of_clusters', 'number_of_cells']].head(5)
| name | cluster_annotation_term_set_name | number_of_clusters | number_of_cells | |
|---|---|---|---|---|
| label | ||||
| CS202210140_476 | Upper-layer intratelencephalic | supercluster | 139 | 455006 |
| CS202210140_477 | Deep-layer intratelencephalic | supercluster | 138 | 228467 |
| CS202210140_473 | Deep-layer near-projecting | supercluster | 72 | 18856 |
| CS202210140_474 | Deep-layer corticothalamic and 6b | supercluster | 116 | 78396 |
| CS202210140_484 | MGE interneuron | supercluster | 211 | 222434 |
Let’s visualize cluster and cells counts for of the classification levels using bar plots.
def bar_plot_by_level_and_type(df, level, fig_width = 8.5, fig_height = 4):
fig, ax = plt.subplots(1, 2)
fig.set_size_inches(fig_width, fig_height)
for idx, ctype in enumerate(['clusters', 'cells']):
pred = (df['cluster_annotation_term_set_name'] == level)
names = df[pred]['name']
counts = df[pred]['number_of_%s' % ctype]
colors = df[pred]['color_hex_triplet']
ax[idx].barh(names, counts, color=colors)
ax[idx].set_title('Number of %s by %s' % (ctype,level)),
ax[idx].set_xscale('log')
if idx > 0:
ax[idx].set_yticklabels([])
plt.show()
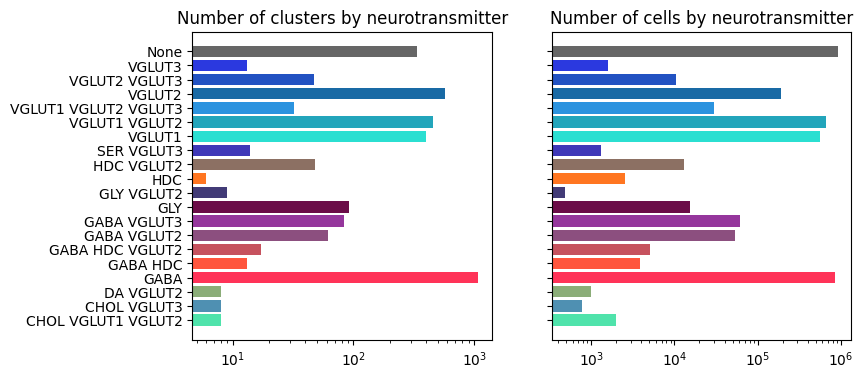
Neurotransmitter cluster and cell counts#
The majority of clusters and cells are of glutamatergic, GABAergic or GABA-glycinergic neurotransmitter types.
df = term_with_counts
level = 'neurotransmitter'
pred = (df['cluster_annotation_term_set_name'] == level)
bar_plot_by_level_and_type(term_with_counts, 'neurotransmitter')
plt.show()
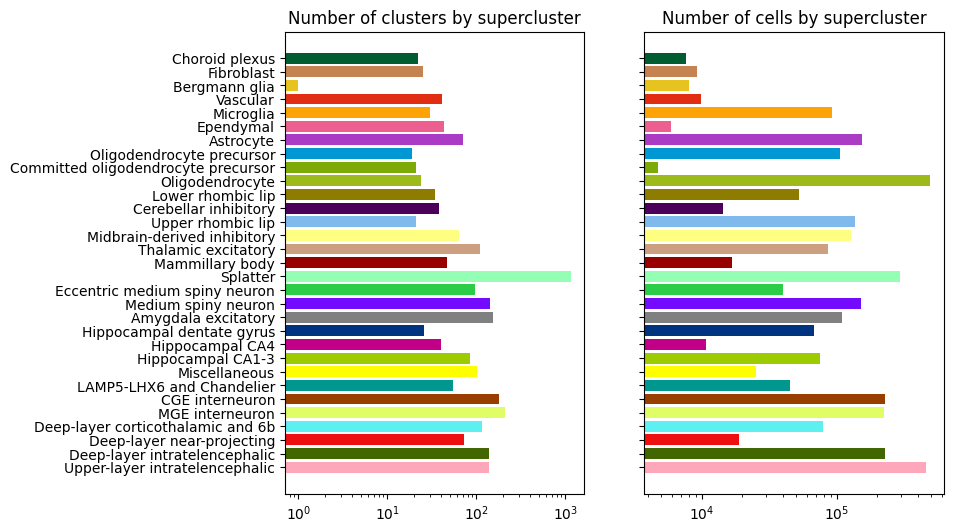
Supercluster level cluster and cell counts#
Supercluster “Splatter” contains the largest number of subclusters (~1k), while “Oligodendrocyte” and “Upper-layer intratelencephalic” have the most cells.
bar_plot_by_level_and_type(term_with_counts, 'supercluster', 8.5, 6)
plt.show()
Visualizing cluster and term distributions#
We can explore the relationship and distribution of clusters between term sets by creating a pivot table using pandas groupby fuunction. Each row of the resulting dataframe represents a cluster, each column represents a term set and the value in the table is the name of the term that has been associated with the cluster for that specific term set.
pivot = membership.groupby(['cluster_alias', 'cluster_annotation_term_set_name'])['cluster_annotation_term_name'].first().unstack()
pivot = pivot[term_set['name']] # order columns
pivot
| cluster_annotation_term_set_name | subcluster | cluster | supercluster | neurotransmitter |
|---|---|---|---|---|
| cluster_alias | ||||
| 0 | URL_297_0 | URL_297 | Upper rhombic lip | VGLUT1 |
| 1 | URL_308_1 | URL_308 | Upper rhombic lip | VGLUT1 |
| 2 | URL_308_2 | URL_308 | Upper rhombic lip | VGLUT1 |
| 3 | URL_308_3 | URL_308 | Upper rhombic lip | VGLUT1 |
| 4 | URL_308_4 | URL_308 | Upper rhombic lip | VGLUT1 |
| ... | ... | ... | ... | ... |
| 3308 | Nkcell_2_3308 | Nkcell_2 | Miscellaneous | None |
| 3309 | Nkcell_2_3309 | Nkcell_2 | Miscellaneous | None |
| 3310 | Nkcell_2_3310 | Nkcell_2 | Miscellaneous | None |
| 3311 | Nkcell_2_3311 | Nkcell_2 | Miscellaneous | None |
| 3312 | Nkcell_2_3312 | Nkcell_2 | Miscellaneous | None |
3313 rows × 4 columns
We can also obtain a cluster annotation color pivot table in the same way.
color = membership.groupby(['cluster_alias', 'cluster_annotation_term_set_name'])['color_hex_triplet'].first().unstack().fillna('#f9f9f9')
color = color[term_set['name']] # order columns
color.columns = ['%s_color' % x for x in color.columns]
color
| subcluster_color | cluster_color | supercluster_color | neurotransmitter_color | |
|---|---|---|---|---|
| cluster_alias | ||||
| 0 | #7E807A | #909FF6 | #80BAED | #2BDFD1 |
| 1 | #C54945 | #A5B0D9 | #80BAED | #2BDFD1 |
| 2 | #5232B7 | #A5B0D9 | #80BAED | #2BDFD1 |
| 3 | #31BEBA | #A5B0D9 | #80BAED | #2BDFD1 |
| 4 | #C8A9BC | #A5B0D9 | #80BAED | #2BDFD1 |
| ... | ... | ... | ... | ... |
| 3308 | #3D84BA | #E2EE30 | #FEFE00 | #666666 |
| 3309 | #C25347 | #E2EE30 | #FEFE00 | #666666 |
| 3310 | #B9BFB4 | #E2EE30 | #FEFE00 | #666666 |
| 3311 | #6CC93D | #E2EE30 | #FEFE00 | #666666 |
| 3312 | #423149 | #E2EE30 | #FEFE00 | #666666 |
3313 rows × 4 columns
For a given pair of term sets A and B, define a function distribution that creates a cluster count table where the rows are terms in term set A, columns are terms in term set B and the table values being the number of clusters that is shared between the terms.
def distribution(A, B):
AxB = pivot.groupby([A, B])[['cluster']].count()
AxB.columns = ['number_of_clusters']
AxB = AxB.unstack().fillna(0)
B_names = [x[1] for x in list(AxB.columns)]
pred = (term['cluster_annotation_term_set_name'] == B)
term_by_name = term[pred].set_index('name')
B_colors = term_by_name.loc[B_names, 'color_hex_triplet']
return AxB, B_names, B_colors
Function stacked_bar_distribution takes the results of distribution as input to create distribution stacked bar plot.
def stacked_bar_distribution(AxB, B_names, B_colors, fig_width = 6, fig_height = 6):
fig, ax = plt.subplots()
fig.set_size_inches(fig_width, fig_height)
bottom = np.zeros(len(AxB))
for i, col in enumerate(AxB.columns):
ax.barh(AxB.index, AxB[col], left=bottom, label=col[1], color=B_colors[i])
bottom += np.array(AxB[col])
ax.set_title('Distribution of %s in each %s' % (AxB.columns.names[1], AxB.index.name))
ax.legend(loc='center left', bbox_to_anchor=(1, 0.5))
plt.show()
return fig, ax
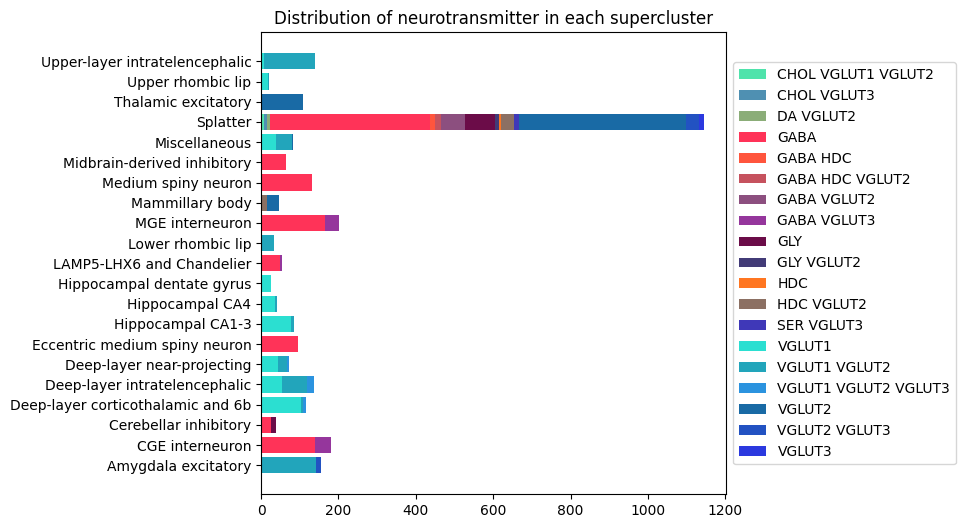
Distribution of neurotransmitter superclusters#
Here is the distribution of neurotransmitter types at the supercluster level. Most contain more than one type with splatter showing the most diversity of neurotransmitter types.
AxB, B_names, B_colors = distribution('supercluster', 'neurotransmitter')
fig, ax = stacked_bar_distribution(AxB, B_names, B_colors, 6, 6)
plt.show()
/var/folders/kc/7glrmt5n67x16yj_tg86t49c0000gp/T/ipykernel_55318/1772253940.py:9: FutureWarning: Series.__getitem__ treating keys as positions is deprecated. In a future version, integer keys will always be treated as labels (consistent with DataFrame behavior). To access a value by position, use `ser.iloc[pos]`
ax.barh(AxB.index, AxB[col], left=bottom, label=col[1], color=B_colors[i])
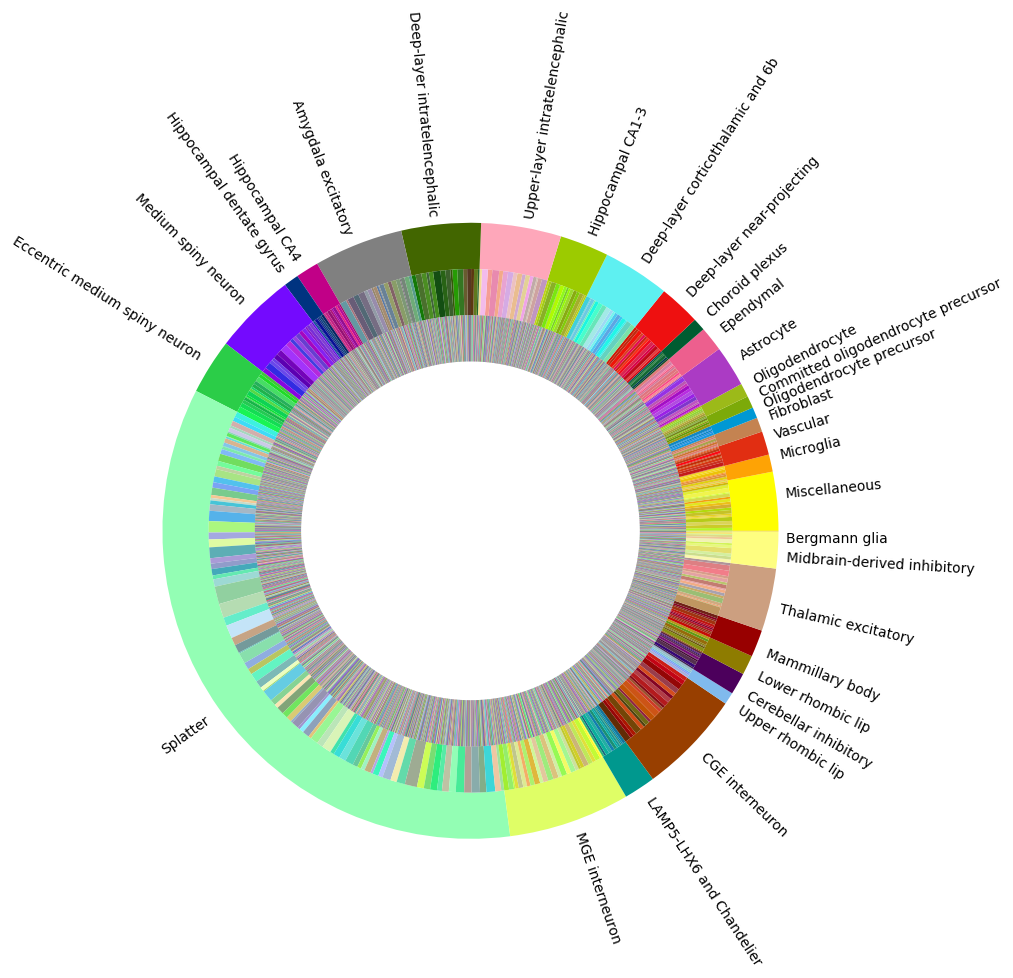
Visualizing the human whole brain taxonomy#
Term sets: supercluster, cluster, and subcluster forms a three level human whole brain taxonomy. We can visualized the taxonomy as a sunburst diagram that shows the single inheritance hierarchy through a series of rings, that are sliced for each annotation term. Each ring corresponds to a level in the hierarchy. We have ordered the rings so that the supercluster level is the outer most ring so that we can add in labels. Rings are sliced up and divided based on their hierarchical relationship to the parent slice. The angle of each slice is proportional to the number of clusters belonging to the term.
levels = ['supercluster', 'cluster', 'subcluster']
df = {}
for lvl in levels :
pred = term_with_counts['cluster_annotation_term_set_name'] == lvl
df[lvl] = term_with_counts[pred]
df[lvl] = df[lvl].sort_values(['parent_term_label', 'label'])
fig, ax = plt.subplots()
fig.set_size_inches(10, 10)
size = 0.15
for i, lvl in enumerate(levels):
if lvl == 'supercluster' :
ax.pie(df[lvl]['number_of_clusters'],
colors=df[lvl]['color_hex_triplet'],
labels = df[lvl]['name'],
rotatelabels=True,
labeldistance=1.025,
radius=1,
wedgeprops=dict(width=size, edgecolor=None),
startangle=0)
else :
ax.pie(df[lvl]['number_of_clusters'],
colors=df[lvl]['color_hex_triplet'],
radius=1-i*size,
wedgeprops=dict(width=size, edgecolor=None),
startangle=0)
plt.show()